10-K: Annual report [Section 13 and 15(d), not S-K Item 405]
Published on April 1, 2026
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
For the fiscal year ended
OR
For the transition period from __________ to __________
Commission file number:
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| N/A |
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.0001 par value per share
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | Smaller reporting company | Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No
The aggregate market value (approximate) of the registrant’s common equity held by non-affiliates based on the most recent sales price of a share of the registrant’s common share on June 30, 2025 (the last business day of the registrant’s most recently completed second fiscal quarter) was $
As of March 26, 2026,
Documents Incorporated by Reference
TABLE OF CONTENTS
i
NOTES
As used in this Annual Report on Form 10-K (this “Report”), unless otherwise stated or the context clearly indicates otherwise, the terms “Adaptin,” the “Company,” “we,” “us” and “our” refer to Adaptin Bio, Inc., incorporated in the State of Delaware, and its subsidiaries after giving effect to the Merger (as defined below) and the company name change described herein.
ii
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Report includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Exchange Act. Forward-looking statements relate to, among others, our plans, objectives and expectations for our business, operations and financial performance and condition, and can be identified by terminology such as “may,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “will,” “could,” “project,” “target,” “potential,” “continue” and similar expressions that do not relate solely to historical matters. Forward-looking statements are based on management’s belief and assumptions and on information currently available to management. Although we believe that the expectations reflected in forward-looking statements are reasonable, such statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by forward-looking statements.
Forward-looking statements include, but are not limited to, statements about:
| ● | our ability to raise additional money to fund our operations for at least the next twelve months as a going concern; |
| ● | our ability to develop our current and any future product candidates; |
| ● | our ability to receive marketing approval from the U.S. Food and Drug Administration (“FDA”) for our product candidates; |
| ● | our ability to maintain our license rights to our intellectual property and to adequately protect or enforce our intellectual property rights; |
| ● | our reliance on third parties to supply drug substance and drug product for our clinical trials and preclinical studies, and produce commercial supplies of product candidates; |
| ● | our ability to market and commercialize our products, if approved; |
| ● | our product candidates’ ability to achieve market acceptance, if approved; |
| ● | developments and projections relating to our competitors and our industry; |
| ● | our ability to adequately control the costs associated with our operations; |
| ● | our dependence on third-party reimbursement for commercial viability; |
| ● | the impact of current and future laws and regulations, especially those related to drug development and drug pricing controls; |
| ● | potential cybersecurity risks to our operational systems, infrastructure, and integrated software by us or third-party vendors; and |
| ● | the development of a market for our Common Stock. |
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, operating results, business strategy, short-term and long-term business operations and objectives, and financial needs. These forward-looking statements are subject to a number of risks, uncertainties, and assumptions. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties, and assumptions, the future events and trends discussed in this Report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
iii
PART I
ITEM 1. BUSINESS.
Our Company
The Company’s objective is to develop and commercialize products utilizing novel technology that enhances the delivery of drugs and other compounds to the brain and other tissues for a variety of indications. Our novel technology was originally developed by researchers in the Department of Neurosurgery at Duke University and licensed by us in 2023.
Our website address is www.adaptinbio.com and we can be contacted at info@adaptinbio.com. Information contained on, or that can be accessed through, our website is not a part of this Report.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or the JOBS Act, and, for as long as we continue to be an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies but not to emerging growth companies, including:
| ● | not being required to have our independent registered public accounting firm audit our internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act; |
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports and annual report on Form 10-K; and |
| ● | exemptions from the requirements of holding non-binding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
Our status as an emerging growth company will end upon the earlier of: (a) the fifth anniversary of the first sale of our common equity securities pursuant to an effective registration statement; (b) the last day of the fiscal year in which we have more than $1.235 billion in annual gross revenues; (c) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities held by non-affiliates; and (d) the date on which we have issued, in any three-year period, more than $1 billion in non-convertible debt securities.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to avail ourselves of this provision of the JOBS Act. As a result, we will not be subject to new or revised accounting standards at the same time as other public companies that are not emerging growth companies. Therefore, our consolidated financial statements may not be comparable to those of companies that comply with new or revised accounting pronouncements as of public company effective dates.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a “smaller reporting company” even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as our voting and non-voting Common Stock held by non-affiliates is less than $250 million measured on the last business day of our second fiscal quarter, or our annual revenues is less than $100 million during the most recently completed fiscal year and our voting and non-voting Common Stock held by non-affiliates is less than $700 million measured on the last business day of our second fiscal quarter.
The Merger
On February 11, 2025, Adaptin Bio, Inc. completed the business combination contemplated by that certain Agreement and Plan of Merger and Reorganization, dated as of February 11, 2025, by and among Unite Acquisition 1 Corp. (“Unite Acquisition”), a public shell company incorporated in the State of Delaware on March 10, 2022, its wholly-owned subsidiary, Adaptin Acquisition Co., a Delaware corporation formed in the State of Delaware on January 30, 2025 (“Merger Sub”), and Adaptin Bio Operating Corporation (formerly “Adaptin Bio, Inc.”) (the “Merger”). Upon the completion of the Merger, Unite Acquisition changed its name to “Adaptin Bio, Inc.”
1
Business Overview
We are a biopharmaceutical company pioneering a transformational approach to enhance the transfer of therapeutics into the brain, facilitating the treatment of brain cancers and other unmet medical conditions. The Company’s proprietary technology harnesses the human immune system’s ability to target, recognize, destroy or deliver therapeutics to specific cells, including cancer cells. Our mission is to be the global leader and pioneer of this new treatment paradigm, integrating recombinant technology, gene therapy and cell therapy to address the challenges of targeting and delivering effective therapies, including to the brain for cancer and other central nervous system (“CNS”) indications.
The cause(s), or etiology, of many diseases can be addressed in part through manipulation of engineered cells. We view targeted manipulation of the human immune system, together with recombinant technology and/or gene therapy, as a therapeutically disruptive transformation in the way we treat brain and other diseases. Our lead product candidate has been recently accepted under an investigator-led Investigational New Drug application (“IND”) to begin first-in-human studies in brain cancer. Assuming success of those studies, our experienced group of scientists and business leaders intend to develop our proprietary in vivo and ex vivo technology platforms to revolutionize treatment across a broad array of other therapeutic areas with unmet treatment needs, including CNS disorders, autoimmune disease and cardiovascular diseases, among others. Pursuing other therapeutic areas will likely require us to either raise a significant amount of additional capital or to engage in strategic transactions such as spin-offs or out-licenses. Our goal is to initiate preclinical studies on additional product candidates in 2027, if not earlier.
Our novel technology was originally developed by researchers in the Department of Neurosurgery at Duke University, led by Dr. John H. Sampson, the prior Robert H. and Gloria Wilkins Distinguished Professor and Chair of the Department of Neurosurgery and currently the Dean and Vice Chancellor at the University of Colorado School of Medicine. The group recognized that adoptive transfer of specifically activated functional human immune cells significantly increases the “hitchhiking” and intracerebral accumulation of macromolecules that are bound to their surface. While circulating naïve T cells do not typically penetrate the CNS, activated T cells are known to traffic frequently past the blood brain barrier (“BBB”) and perform routine immune surveillance in the CNS. Adaptin and its collaborators at Duke University are taking advantage of this CNS trafficking to enhance the localization of macromolecules and other agents to the CNS for cancer and other CNS disorders.
We are closely working with researchers at Duke University to translate preclinical proof of concept data of its first proprietary platform technology called BRiTE (Brain Bispecific T-cell Engager) into human clinical trials. BRiTE focuses on the transport of difficult to deliver T cell targeting agents to tumor tissue, including in the immunoprivileged brain and overcoming the challenges with other immunotherapeutic approaches. BRiTE is a translatable method to specifically target malignant glioma using a tumor-specific, fully human bispecific antibody that redirects patients’ own T cells to recognize and destroy tumor cells.
The first application of our technology is APTN-101, a proprietary epidermal growth factor receptor variant III, or EGFRVIII, BRiTE in order to eliminate malignant glioma tumors in a variety of aggressive preclinical tumor models where the tumor is implanted behind the BBB in the CNS (i.e., orthotopic). We designed APTN-101 to specifically redirect T cells against tumors expressing a well-characterized, mutated form of epidermal growth factor receptors (“EGFRs”) known as EGFRVIII, on a number of tumor types, including glioblastoma, breast and lung cancer. Because EGFRVIII is exclusively expressed on tumor cells, but not normal healthy cells, we believe it represents an ideal target for immunotherapy. We have made significant progress towards first-in-human clinical studies, including:
| ● | A pre-IND meeting with the FDA outlining a clear path to filing an IND; |
| ● | Completion of single-dose IND-enabling preclinical studies; |
2
| ● | Submission in April 2023 of an IND for an investigator-initiated, single-dose clinical trial, and its acceptance in May 2023 by the FDA; and |
| ● | Manufacturing of APTN-101 in more than sufficient quantities for Phase 1 trials. |
| ● | Submission in August 2025 of an IND amendment to include multiple dosing for the investigator-initiated, multi-dose clinical trial, and its acceptance in October 2025 by the FDA; and |
| ● | Received both internal and external Institutional Review Board (IRB) approval for the investigator-initiated, multi-dose clinical trial evaluating APTN-101 in the treatment of glioblastoma multiforme (GBM) in March 2026. |
We also expect to expand our proprietary platform to other targets and indications. The Company is exploring several external opportunities to continue to advance and expand the product pipeline.
Strategy
Our goal is to become a leading biopharmaceutical company focused on the transfer of drugs across barriers and to targeted tissues, including the brain and CNS, to transform current treatment paradigms for patients and address unmet medical needs. The critical components of our strategy are as follows:
| ● | Advance the development of APTN-101 for the treatment of glioblastoma. The FDA’s acceptance in May 2023 of the IND for APTN-101 and the acceptance in October 2025 of the amended IND for APTN-101 for the multi-dose first-in-human clinical trial in glioblastoma patients. |
| ● | Advance preclinical development of APTN-101 to support one or more additional INDs for additional kinds of cancer. We have designed APTN-101 to incorporate EGFRvIII, which is expressed on a number of tumor types, including breast and lung cancer (with or without brain metastases), so we are considering pre-clinical work to support INDs for these indications to be filed. |
| ● | Design and advance other early-stage drug product candidates for undisclosed rare and unmet needs. Because our proprietary technology enables drugs to cross barriers and target tissues, including the brain, we believe it has numerous potential applications in areas of unmet medical need. We are evaluating which of those indications would be most strategic to pursue in the near-term. |
| ● | Acquire, in-license or develop complementary delivery technologies that will allow us to produce BRiTE compounds or manipulate and activate immune cells in vivo. We continually evaluate technologies that will further enhance therapeutic effect, improve safety and manufacturability, or reduce costs of our products. |
| ● | Acquire targeted clinical compounds for conditions with unmet needs where our technology could be transformative. We continually evaluate development and in-licensing opportunities and may acquire clinical compounds for conditions with unmet medical needs where our technology’s ability to cross barriers and target specific tissues, including the brain, could be transformative of the treatment paradigm. |
| ● | Pursue a capital-efficient commercialization strategy. For products with smaller and/or orphan patient populations, our plan is to build an infrastructure to commercialize our drug products within the United Sates. Drawing upon our experience in commercializing specialty pharmaceutical products, we aim to build a specialized yet efficient infrastructure that will support the entire commercialization continuum, including stakeholder education, treatment decision and initiation, and product access throughout the patient journey. In addition, we plan to seek established companies to commercialize our drug products for larger addressable markets and outside of the United States. |
| ● | Leverage, protect and enhance our intellectual property portfolio and secure patents for additional products and indications. We intend to expand our intellectual property, grounded in securing composition of matter and method of use patents for new products and indications. We plan to enhance the intellectual property portfolio further through learnings from ongoing preclinical studies, clinical trials and manufacturing processes. |
| ● | Outsource capital-intensive operations. We plan to continue to outsource capital-intensive operations, including most clinical development and all manufacturing operations of our product candidates, and to facilitate the rapid development of our pipeline by using high quality specialist vendors and consultants in a capital efficient manner. |
3
Glioblastoma
Background
Glioblastoma multiforme (“GBM”), the highest grade (World Health Organization (“WHO”) grade IV) astrocytoma, is the most common and malignant brain tumor, accounting for about 50% of all gliomas and 12%-15% of all brain tumors. GBM tumor cells, which arise from stem cells or immature astrocytes due to genetic abnormalities, grow rapidly and disseminate in the brain. In addition, GBM cells can invade the intracranial blood vessels to areas away from the tumor core.
Although glioblastoma can happen anywhere in the brain, it usually forms in the frontal lobe and the temporal lobe. Glioblastoma rarely occurs in the brain stem or spinal cord. As glioblastoma grows, it spreads into the surrounding brain. This makes it difficult to remove the entire tumor with surgery. Although radiation therapy and chemotherapy can reach the tumors, glioblastoma cells can survive and regrow. Glioblastoma is very challenging to treat due to tumor-specific features, such as its rapid growth rate, the poor function of the immune system cells within the tumor, and inherent resistance of the tumor cells to many types of treatments.
There is no direct risk factor associated with most cases of glioblastoma. Certain rare genetic diseases, such as Li-Fraumeni and Lynch syndrome, are associated with gliomas. However, these affect only a small portion of patients with glioblastoma. Besides genetic syndromes, the only well-established risk factor is prior exposure to ionizing radiation that is used to treat certain head and neck cancers.
Brain tumor symptoms vary and depend on the tumor location. The most common glioblastoma symptoms are headaches, seizures, and progressively worsening numbness or weakness. Headaches with red flag symptoms warrant a trip to the doctor for a neurologic evaluation. Red flag symptoms include waking up due to pain, worsening pain on changing position, and continuous pain not relieved with over-the-counter headache medications.
Neurologic imaging with an MRI of the brain is often the first step in diagnosis. Brain imaging showing contrast-enhancing masses can be suggestive of glioblastoma. Most cases can be definitively diagnosed after surgery through histological testing. This takes place when a neuropathologist examines tissue or cells under a microscope to help confirm a glioblastoma diagnosis.
Incidence and Mortality
In the United States, the average annual age-adjusted incidence of glioblastoma is 3.2 per 100,000 population (or about 12,000 patients annually) with an average age of 64 at diagnosis. Glioblastoma is 1.6 times more common in males compared with females and 2.0 times higher in Caucasians compared to African Americans, with lower incidence in Asians and American Indians. Globally, glioblastoma incidence is highest in North America, Australia, and Northern and Western Europe. Veterans who served in Iraq or Afghanistan are 26% more likely to develop glioblastoma, according to the U.S. Department of Veterans Affairs and National Institutes of Health data, likely due to environmental exposures. Malignant primary brain tumors are the most frequent cause of cancer death in children, are more common than Hodgkin lymphoma, ovarian and testicular cancer and are responsible for more deaths than malignant melanoma.
Overall, the one-year relative survival rate is about 40% for patients diagnosed in the United States. The five-year survival rate is only about 5%. Treatment outcome remains poor, with a median survival rate of about 15 months.
Current Treatment/Management
A patient’s care team will take into account age, functional status, medical history and medication tolerability when planning the best treatment. For most newly diagnosed patients, the standard approach utilizes what is known as the Stupp protocol. Treatment is comprised of maximal surgical resection, which allows for accurate histological diagnosis, tumor genotyping, and a reduction in tumor volume, followed by 6 weeks of radiotherapy and concomitant daily temozolomide and a further 6 cycles of maintenance temozolomide. In patients with minimal functional impairment, the median overall survival (“OS”) is 15 months for radiotherapy plus temozolomide versus 12 months for radiotherapy alone.
4
Treatment options in the relapsed or recurrent setting are less well defined, with no established standard of care and little evidence for any interventions that prolong OS. Indeed, a significant proportion of patients may not even be eligible for second-line therapy. Options include further surgical resection, reirradiation, systemic therapies such as carmustine or bevacizumab, combined approaches, or supportive care alone.
Patients may also be treated with tumor treatment fields. This is a portable device placed on the scalp that uses mild electrical fields to try to interrupt cancer cell growth.
Standard-of-care treatments fail to specifically eliminate tumor cells and are limited by incapacitating damage to surrounding normal brain and systemic tissues leading to lymphopenia and many other detrimental side effects. All of this demonstrates that a more targeted immunotherapeutic approach is needed. Over the last decade, emerging immunotherapies (such as monoclonal antibodies, oncolytic virus therapy, adoptive cell therapy, and cellular vaccines therapy) aimed at improving specific immune response against tumor cells have brought a glimmer of hope to patients with GBM. Adoptive cell therapy, including tumor-infiltrate lymphocytes (“TILs”) transfer and genetically engineered T cells transfer, is one of the most significant breakthroughs in the field of immune-oncology. Chimeric antigen receptor (“CAR”) engineered autologous T cells have produced sustained remissions in refractory lymphomas, but this approach needs further study in the treatment of solid tumors. While there is significant potential with targeted immunotherapy in GBM, significant challenges remain, including primarily the difficulty of crossing the BBB.
Bi-Specific Antibodies
The concept of bispecific antibodies was first introduced in 1980s as a method to target multiple antigens by a single antibody. The recombinant bispecific antibodies are classified into two types. The first are antibodies containing the crystallizable region (i.e., Fc-containing antibodies) and the second are antibody derivatives without Fc regions. The Fc region is the tail region of an antibody that interacts with cell surface receptors called Fc receptors and some proteins of the complement system. The mechanism of action of bispecific antibodies includes binding to the tumor cells on one side through the Fab (antigen-binding region) portion of the antibody against the tumor-specific antigen (such as CD19, HER2, EGFR, or GD2) and to the immune effector cells such as T cells and NK cells, which leads to activation of those immune effector cells and Fc-receptor bearing phagocytic cells such as monocytes/macrophages that can also mediate direct lysis of the tumor cells.
Bispecific antibodies termed bispecific T cell engagers (“BiTEs”) are monomeric proteins consisting of two antibody-derived single-chain variable fragments (“scFvs”) translated in tandem. These constructs possess one effector-binding arm specific for the epsilon subunit of T-cell CD3 and an opposing target-binding arm directed against an antigen that is expressed on the surface of tumor cells (e.g., EGFRvIII).
We believe EGFRvIII is an attractive target tumor specific antigen, in part because it is specific to cancer cells and is not expressed in non-tumor tissue. Importantly, antibodies directed against EGFRvIII are entirely tumor-specific and do not cross react with the wild-type receptor located on healthy cells. Therefore, by retargeting T cells against the tumor-specific EGFRvIII antigen, we believe we can avoid killing healthy tissue and the related adverse effects. EGFRs are involved in deregulated cancer signaling pathways, leading to atypical proliferation and growth of tumor cells. EGFRvIII is the most common variant not presented in a major histocompatibility complex (“MHC”) -dependent manner and is seen in approximately 31 to 50% of patients with GBM and in a broad array of other cancers including breast and lung carcinoma. Lung and breast carcinoma are the two main types of cancer that lead to secondary brain tumors (i.e., brain metastases) in about 25% of these patients. Among patients with EGFRvIII-positive GBM, 37 to 86% of tumor cells express the mutated receptor, indicating that the mutation is translated with significant consistency.
5
Among patients with GBM, expression of EGFRvIII is an independent, negative prognostic indicator. EGFRvIII also enhances the growth of neighboring EGFRvIII-negative tumor cells via cytokine-mediated paracrine signaling and by transferring a functionally active oncogenic receptor to EGFRvIII-negative cells through the release of lipid-raft related microvesicles. Recent research has also found that EGFRvIII is expressed in glioma stem cells, an important consideration given the paradigm that tumor stem cells represent a subpopulation of cells that give rise to all differentiated tumor cells. Altogether, the specificity, high frequency of surface expression and oncogenicity of the EGFRvIII mutation make it an ideal target for antibody-based immunotherapy.
The divalent structure of BiTEs brings T cells into close proximity to the tumor cell, creating a synapse. Following BiTE-mediated synapse formation, T cells proliferate, secrete pro-inflammatory cytokines and express surface activation markers. Following BiTE-mediated synapse formation, T cells release perforin and granzyme proteases that kill tumor cells. BiTEs are capable of mediating serial rounds of killing and can trigger specific tumor cell killing from naïve T cells at exceedingly low concentrations and effector-to-target ratios.
It is well established that certain gliomas, such as glioblastoma, are uniquely shielded from the immune system due to its location within the CNS. While this privilege is not absolute, a significant proportion of tumors have been noted to be devoid of any TILs that can be redirected by bispecific T-cell engagers. In those tumors that do demonstrate invasion by TILs, they are often induced to be dysfunctional and anergic by the suppressive tumor microenvironment. Increased numbers of intratumoral CD8+ cytotoxic T lymphocytes (“CTLs”) have been associated with favorable outcomes in patients with glioblastoma.
Concomitant administration of stimulated CTLs may therefore synergistically enhance the efficacy of this treatment. The migration of T-cell engagers across the BBB may also be facilitated by activated T cells which adhere to the brain microvascular endothelium and subsequently cross by diapedesis. Concurrent administration of activated functional T cells could therefore enhance the trafficking of bispecific T-cell engagers and other therapeutics into the intracranial compartment, increasing their density at the tumor site and thus the therapeutic effect.
Additionally, target cell killing with BiTE occurs in the absence of regular MHC peptide antigen recognition and costimulation and is therefore resistant to certain immune escape mechanisms affecting antigen presentation and those affecting generation of tumor-specific T cell clones. Because the CD3ε target of BiTE antibody construct is the same in CD8+ and CD4+ T cells of any phenotype, they are all engaged, leading to a polyclonal T cell activation, expansion and broad tumor cell killing.
Bispecific T-cell engagers offer immunotherapy in a manufacturing format which is both scalable and standardizable. In contrast to CAR T cells, T-cell engagers do not require initial lymphodepletion. Ex vivo manipulation of autologous cells has significant limitations, including the need for a centralized manufacturing infrastructure with extensively trained laboratory personnel to genetically modify each patient’s own T cells, use viral transduction which poses uncertain risks, are limited to the initial subset of T cells manipulated and infused, and still face uncertainty as to the optimal T cell phenotype to infuse.
Our Product Pipeline
APTN-101
We have recently reported the development of our first novel T cell engaging molecule, known as APTN-101, using our BRiTE technology. BRiTE focuses on the transport of difficult to deliver T cell targeting agents across the BBB allowing access to the immunoprivileged brain and overcoming the challenges with other immunotherapeutic approaches. This is accomplished by sequentially or simultaneously administering both BRiTE and specifically activated T cells by adoptive transfer. APTN-101 was designed to specifically redirect T cells against tumors expressing a well-characterized, mutated form of the EGFR, EGFRvIII, on a number of tumor types, including GBM. Because EGFRvIII is exclusively expressed on tumor cells, but not normal healthy cells, it represents an ideal target for immunotherapy.
APTN-101 (EGFRvIII x CD3 BRiTE) successfully activates human T cells against EGFRvIII expressing target cells, in the absence of any additional immunostimulatory signal, resulting in the secretion of Th-1-associated cytokines and tumor-cell killing. APTN-101 is similarly effective in vivo. Intravenous administration of APTN-101 induced consistent antitumor responses in mice bearing established, late-stage, aggressive, intracerebral patient-derived gliomas, rapidly achieving complete remission rates as high as 75% in the absence of apparent toxicity. Given the exquisite tumor-specificity of APTN-101, it represents a critical conceptual advance in safety contrary to target antigens having a promiscuous expression pattern.
6
The concept of BRiTE is the combination of novel T cell targeting agents with specifically activated polyclonal T cells. In order to exert antineoplastic affects against brain tumors, both the T cell targeting agent and T cells need to efficiently access areas that have long been considered as immunoprivileged. While circulating naïve T cells do not typically penetrate the CNS, activated T cells are known to cross the BBB to perform routine immunosurveillance of the central nervous system (Figure 1).
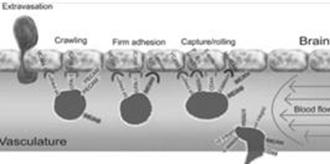
Figure 1- The process of T cells crossing the inflamed BBB is coordinating and sequential. Briefly, activated T cells initially arresting on the endothelium is mediated by the lymphocyte-associated antigen-1 (“LFA- 1”) and α4β1-integrin expressed on the T cells, respectively binding to the intracellular cell adhesion molecule 1 (“ICAM1”) and adhesion molecules vascular cell adhesion molecule 1 (“VCAM1”) on brain endothelial cells. Subsequently, the T cell crawling and polarization exclusively involve LFA-1 and ICAM1/2 interactions. After arriving at sites where are rich in the laminin isoform α4 but not laminin β5, the T cells use α6β1-integrin to traverse the endothelial basement membrane.
Upon intravenous administration, the T cell targeting agent (i.e., EGFRviii x CD3) binds to circulating T cells via its CD3 receptor and carries or “hitchhikes” the agent to tumors located behind the BBB. Studies in aggressive orthotopic GBM models have revealed that adoptive transfer of activated T cells significantly increases the biodistribution of intravenously administered EGFRvIII x CD3 BRiTE to orthotopic glioma (Figure 2).
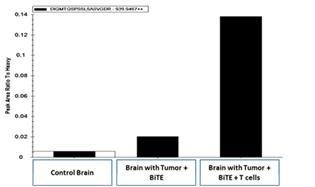
Figure 2- Mass spectroscopy demonstrates that pre-administration (four days) of ex vivo activated T cells increases the biodistribution of intravenously administered EGFRvIII x CD3 to the brain parenchyma.
BRiTE circumvents ordinary clonotypic T-cell specificity, potentially allowing any T-cell, regardless of endogenous specificity or phenotype, to exert an anti-neoplastic effect. In vivo experiments show that BRiTEs can reactivate potentially unresponsive, anergic T cells, such as those frequently encountered among TIL populations thereby enhancing the spread of T cells reactive towards other antigens (epitope spreading). Proximal contact between T cells and tumor cells could directly reactivate tumor infiltrating lymphocytes specific for cancer antigens other than directed by BRiTE, without cross-presentation. The EGFRvIII x CD3 BRiTE molecule has the potential to directly activate and expand pre-existing T cells among a polyclonal population that are specific for tumor antigens other than EGFRvIII. Indeed, others have discovered that by re-activating pre-existing T cell clones using a CD3 binding bispecific antibody specific for Wilms’ tumor protein (WT1) it is possible to induce effective and persistent epitope spreading responses to multiple antigens. If the clinical utility of this mechanism of epitope spreading is confirmed, this could provide an exciting mechanism to combat tumor heterogeneity.
7
Importantly, our data suggest that once APTN-101 reaches the brain, it can activate even suppressive regulatory T-cells (“Tregs”) to kill glioblastoma tumor cells by redirecting their natural granzyme-mediated cytotoxic potential and enhance a cytotoxic immune response. These findings not only highlight a new mechanism by which BRiTEs may circumvent certain aspects of Treg mediated suppression, but also have broader implications with regard to the natural functional role of activated, tumor infiltrating Tregs that ordinarily suppress and kill cytotoxic T lymphocytes in the tumor microenvironment.
By tethering cytotoxic effectors to target cells without the need for antigen presentation via the MHC, BRiTEs can furthermore overcome tumor immune escape mechanisms, such as the downregulation of MHC.
In addition to enhancing the biodistribution of EGFRvIII x CD3 BRiTE to the brain, the activated polyclonal T cells (compared to the addition of no activated polyclonal T cells or naïve polyclonal T cells) have the potential to restore effector T cells function at intracerebral sites (Figure 3) leading to cures in greater than 75% of animals.
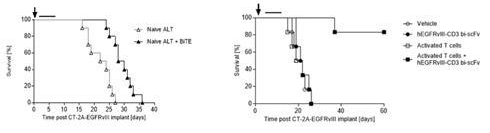
Figure 3- IV administration of activated T cells enhances hEGFRvIII-CD3 bi-scFv efficacy against syngeneic, highly-invasive, orthotopic glioma (right panel) compared to IV administration of naïve T cells when combined with hEGFRvIII-CD3 bi-scFv (left panel). The highly invasive murine glioma CT-2A-EGFRvIII was implanted orthotopically in human CD3 transgenic mice (females, 8-10 weeks old, n=10 per group).
Next, we hypothesized that the increase in efficacy observed with the pre-administration of activated polyclonal T cells would be abrogated if those T cells were to be blocked from entering the CNS parenchyma. Natalizumab, a clinically approved drug for the treatment of multiple sclerosis, functions by binding to polyclonal T cells and preventing their association with receptors involved in the process of extravasation. Remarkably, in cohorts of mice receiving adoptive transfer of activated polyclonal T cells along with treatment with the extravasation blocking molecule natalizumab, efficacy was decreased to levels observed in cohorts that did not receive adoptive transfer of activated polyclonal T cells (Figure 4).
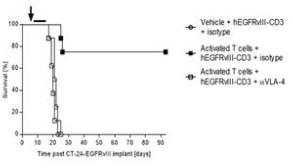
Figure 4- Blocking T cell extravasation with Natalizumab (αVLA-4) abrogates the observed increase in efficacy with adoptive cell transfer (n=8 per group). Natalizumab is a clinically approved drug for the treatment of multiple sclerosis that functions by blocking T cell extravasation.
8
We have also demonstrated an effective “antidote” for any potential toxicity that may result from administration of the EGFRvIII x CD3 BRiTE in a clinical setting. By administering a short peptide that spans the EGFRvIII mutation (PEPvIII), we have effectively blocked bispecific antibody function both in vitro and in vivo, providing a tool highly likely to aid in safe clinical administration of any EGFRvIII targeted bispecific antibody.
The above data demonstrates that BRiTE technology has the ability to transport difficult to deliver agents, including T cell targeting agents, across the BBB and demonstrate superior antineoplastic activity in aggressive orthotopic models of GBM while having an acceptable safety profile. This newly uncovered hitchhiking mechanism of drug delivery to the CNS provides an important tool to enhance the immunotherapy of brain tumors and has potentially far-reaching consequences for the treatment of other CNS disorders, such as Alzheimer’s or Parkinson’s disease, where issues regarding drug delivery to the CNS are relevant.
In summary, the results of preclinical studies demonstrate that the EGFRvIII targeting BRiTE may provide a safe, highly effective therapeutic option for GBM patients. Future studies will determine whether these results can be recapitulated in the clinical setting and whether BRiTEs favorably interact with other therapies that are currently employed as a standard-of-care for GBM patients.
APTN-101 Clinical Studies
The proposed Phase 1 study will evaluate a novel hEGFRvIII-CD3-biscFv Bispecific T cell engager (BRiTE) in patients diagnosed with pathologically documented supratentorial WHO grade IV malignant glioma with an EGFR mutation (either newly diagnosed or at first progression/recurrence) at the Preston Robert Tisch Brain Tumor Center at Duke University.
The primary objective of this Phase 1 study is to determine the safety and tolerability of BRiTE and recommend Phase 2 dose of BRiTE injected with and without activated polyclonal T-cells in among a patient population that includes newly diagnosed patients after completion of standard of care therapy consisting of radiation and adjuvant temozolomide, and patients at first progression (defined as progression during or after standard of care radiation and adjuvant temozolomide).
Another secondary objective is to describe the pharmacokinetics (“PK”) in subjects treated with BRiTE. A population PK analysis will be performed to characterize the PK of BRiTE using the software Nonlinear Mixed Effects Modeling (NONMEM, version 7.2). Different structural PK models (e.g., 1 and 2 compartment) with linear or non-linear (e.g., Michaelis-Menten) kinetics will be fitted to the plasma concentration-time data of BRiTE.
Exploratory objectives include an evaluation of the pharmacodynamics effect of BRiTE, an evaluation of the formation and incidence of anti-BRiTE antibodies, and a description of overall survival and progression-free survival.
Our Intellectual Property
We strive to protect the proprietary technology that we believe is important to our business, including our product candidates and our processes. We seek patent protection in the United States and internationally for our product candidates, their methods of use and processes of manufacture, and any other technology to which we have rights, as appropriate. Additionally, we have licensed the rights to intellectual property related to certain of our product candidates, including patents and patent applications that cover the products or their methods of use or processes of manufacture. The terms of the licenses are described below under the heading “License Agreement.” We also rely on trade secrets that may be important to the development of our business.
We hold a world-wide exclusive license to three issued or allowed United States patents and one pending Patent Cooperation Treaty (“PCT”) patent application covering the enhanced delivery of drugs and other compounds to the brain and other tissues. The patents and patent applications that we licensed provide patent terms or anticipated patent terms ranging from 2031 to 2039 without patent term extensions.
9
Our success will in part depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to our business, the validity and enforceability of our patents, the continued confidentiality of our trade secrets, and our ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology and products.
License Agreement
Patent License Agreement with Duke University
Effective January 11, 2023, we entered into a patent license agreement (the “Duke License”) with Duke University, a nonprofit educational and research institution organized under the laws of North Carolina (“Duke University”), whereby Duke University granted us an exclusive license with a right to grant sublicenses to “BISPECIFIC EGFRvIII ANTIBODY ENGAGING MOLECULES,” “HUMAN BISPECIFIC EGFRvIII ANTIBODY AND CD3 ENGAGING MOLECULES,” “CERTAIN IMPROVED HUMAN BISPECIFIC EGFRvIII ANTIBODY ENGAGING MOLECULES” and “ENHANCED DELIVERY OF DRUGS AND OTHER COMPOUNDS TO THE BRAIN AND OTHER TISSUES” (together, the “precision medicine technology”). With this technology, which we assumed pursuant to the Merger, we intend to develop our BRiTE Platform, a combination of an immune cell engager with activated functional T-cells, which focuses on transporting difficult to deliver agents across the blood brain barrier, allowing access to the immunoprivileged central nervous system. Under the Duke License, we are required to use commercially reasonable efforts to obtain and retain the relevant governmental approvals and to commercialize the precision medicine technology. We must also use reasonable efforts to reach certain commercialization and research and development milestones as outlined in the Duke License.
On August 8, 2024, we entered into a Sponsored Research Agreement (the “2024 SRA”), which we assumed pursuant to the Merger, whereby Duke University agreed to perform research exploring the administration methods of our BRiTE Platform for a fixed fee. The Duke License has been amended (the “First Amendment”) such that we have the option to add any invention conceived as a result of the performance under the 2024 SRA to the license.
On February 18, 2025, we entered into an additional Sponsored Research Agreement (the “2025 SRA”), whereby Duke University agreed to perform research to support the development of GMP manufacturing procedures for the APTN-101 cell product to be infused with the hEGFRvIII-CD3 bi-scFv product as part of the BRiTE study, and for the validation of those procedures in a GMP environment at Duke University’s Marcus Center for Cellular Cures.
As part of the consideration for the license, we issued Duke University shares, representing 5% of our then issued and outstanding common stock on a fully diluted basis. Following the Merger and additional issuances of common stock in 2025, Duke University holds approximately 1.2% of our shares of common stock on a fully diluted basis. We also agreed to make payments based on clinical and commercial milestones, continuing royalty fees on any sales made after approval by regulatory authorities and minimum annual royalty payments that began in 2025. These milestones include initiation of a Phase II or Phase III clinical trials, submission of applications for market approval in multiple jurisdictions including the United States, European Union and Japan and the initiation of post-approval commercial sales in the same jurisdictions. Based on an assumption that all milestones related to the current development program are met during the course of the Duke License, these milestone payments would total approximately $11.7 million. Under the terms of the Duke License, we must pay running royalties equal to low- to mid-single digit percentages of annual net sales, depending on the level of sales by us, our sublicensees and affiliates in that year, and subject to downward adjustment to low single digit percentages of our net annual sales in the event there is no valid claim of a patent for the product, with minimum annual royalty levels established. We also must pay Duke University low to mid-double-digit percentages of any sublicensing fees as set forth in the Duke License. We will be responsible for all patent expenses incurred by Duke University and have reimbursed Duke University approximately $327,000 for patent expenses incurred, prior to the license, by Duke University for filing and prosecution of the patent rights. Additionally, under the terms of the Duke License, we have reimbursed Duke University for patent expenses incurred of approximately $55,000 and $31,000 for the years ended December 31, 2025 and 2024, respectively.
10
The term of the Duke License will extend until the expiration of the last to expire patent rights, subject to early termination as set forth in the Duke License. The foregoing descriptions of the Duke License, First Amendment and SRAs do not purport to be complete and are qualified in their entirety by the terms and conditions of the Duke License, First Amendment and SRAs.
Manufacturing and Supply
We contract with third parties for the manufacturing of all of our product candidates, and for pre-clinical and clinical studies and intend to continue to do so in the future. We do not own or operate any manufacturing facilities and we have no plans to build any owned clinical or commercial-scale manufacturing capabilities. We believe that the use of contract manufacturing organizations (“CMOs”) eliminates the need to directly invest in manufacturing facilities, equipment and additional staff. Although we rely on contract manufacturers, our personnel and consultants have extensive manufacturing experience overseeing CMOs.
We have produced the EGFRvIII x CD3 BRiTE in a fashion suitable for clinical translational and compatible with clinical biologic manufacturing infrastructure. This has included generating and certifying a Master Cell Bank and developing a scalable expression and tag-free purification and formulation process suitable for clinical translation. On the basis of our data and development work, we have had a CMO produce clinical-grade EGFRvIII x CD3 BRiTE suitable for clinical study.
As we further develop our product candidates, we expect to consider secondary or back-up manufacturers for both active pharmaceutical ingredient and drug product manufacturing. To date, our third-party manufacturers have met the manufacturing requirements for our product candidates in a timely manner. We expect third-party manufacturers to be capable of providing sufficient quantities of our product candidates to meet anticipated full-scale commercial demand but we have not assessed these capabilities beyond the supply of clinical materials to date. We currently engage CMOs on a “fee for services” basis based on our current development plans. We plan to identify CMOs and enter into longer-term contracts or commitments if and as we move our product candidates into Phase 3 clinical trials.
We believe alternate sources of manufacturing will be available to satisfy our clinical and potential future commercial requirements; however, we cannot guarantee that identifying and establishing alternative relationships with such sources will be successful, cost-effective, or completed on a timely basis without significant delay in the development or commercialization of our product candidates. All of the vendors we use are required to conduct their operations under current Good Manufacturing Practices (“cGMP”), a regulatory standard for the manufacture of pharmaceuticals.
Competition
The pharmaceutical industry is highly competitive and characterized by intense and rapidly changing competition to develop new technologies and proprietary products, particularly in some of the areas of high unmet medical need that we are targeting. Our potential competitors include both major and specialty pharmaceutical and biotechnology companies worldwide, many of which have far greater resources and access to capital than we do. In particular, Context Therapeutics, Vir Biotechnology and Janux Therapeutics, Inc. are studying immune cell engagers with different targets, and Amgen Inc. and Genentech, Inc. (a wholly owned subsidiary of Roche Holding AG) have programs using a form of EGFRvIII x CD3 bispecific T cell engager (although neither are using them in combination with activated T cells). Our success will be based in part on our ability to identify, develop, and manage a portfolio of safe and effective product candidates that address the unmet needs of patients before our competitors.
Government Regulations
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of drugs, such as those we are developing. Along with our third-party contractors, we will be required to navigate the various preclinical, clinical, and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
11
FDA Regulation of Drugs
Before any of our drug product candidates may be marketed in the United States, they must be approved by the FDA. The process required by the FDA before drug product candidates may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin and must be updated annually or when significant changes are made; |
| ● | approval by an independent IRB or ethics committee for each clinical site before a clinical trial can begin; |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed product candidate for its intended purpose; |
| ● | preparation of and submission to the FDA of a New Drug Application (“NDA”) after completion of all required clinical trials; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA to file the application for review; |
| ● | satisfactory completion of an FDA Advisory Committee review, if required by the FDA; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with cGMP, and to assure that the facilities, methods, and controls are adequate to preserve the product’s continued safety, purity and potency, and of selected clinical investigational sites to assess compliance with current Good Clinical Practices; and |
| ● | FDA review and approval of the NDA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually and when significant changes are made. |
The testing and approval processes require substantial time, effort, and financial resources and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our product candidates. The FDA may delay or refuse approval of an NDA if applicable regulatory criteria are not satisfied, or may require additional testing, information, and/or post-marketing testing and surveillance to monitor safety or efficacy of a product candidate.
If regulatory approval of a product candidate is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA with a Risk Evaluation and Mitigation Strategy (“REMS”) plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries, and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our product candidates under development.
12
FDA Programs for Expedited Review and Increased Exclusivity
A sponsor may seek approval of a product candidate under Fast Track and Breakthrough Therapy programs designed to accelerate the FDA’s review and approval of new drug candidates that meet certain criteria, and/or to receive increased exclusivity under the orphan drug program. We intend to pursue these programs where our product candidates qualify.
Fast Track. A new drug candidate is eligible for Fast Track designation if it is intended to treat a serious or life-threatening condition, fill an unmet medical need, and demonstrate a significant improvement in the safety or effectiveness in the treatment of that condition.
A drug that receives Fast Track designation is eligible for the following:
| ● | more frequent meetings with FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval; |
| ● | more frequent written correspondence from FDA about the design of clinical trials; |
| ● | priority review to shorten the FDA review process for a new drug from ten months to six months; and |
| ● | rolling review, which means we can submit completed sections of its NDA for review by FDA, rather than waiting until every section of the application is completed before the entire application can be reviewed. |
Under the accelerated approval program, the FDA may approve an NDA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Fast Track designation and priority review do not change the standards for approval but may expedite the development or approval process.
Breakthrough Therapy. A new drug candidate is eligible for Breakthrough Therapy designation if it is intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on one or more clinically significant endpoints.
A drug that receives Breakthrough Therapy designation is eligible for the following:
| ● | All Fast Track designation features; |
| ● | Intensive guidance from the FDA on an efficient drug development program, beginning as early as Phase 1; and |
| ● | FDA organizational commitment involving senior managers. |
Orphan Drug Designation. Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug candidate intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that costs of research and development of the drug for the indication can be recovered by sales of the drug in the United States. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Although there may be some increased communication opportunities, orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
13
If a drug candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in very limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
As in the United States, designation as an orphan drug for the treatment of a specific indication in the European Union must be made before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
The inability to obtain or failure to maintain adequate product exclusivity for our product candidates could have a material adverse effect on our business prospects, results of operations and financial condition.
Other Healthcare Laws and Compliance Requirements
Our sales, promotion, medical education, clinical research, and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to the FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services (“CMS”), the U.S. Department of Health and Human Services (“DHHS”) Office of Inspector General, and other divisions of DHHS, and state and local governments.
Our business and our relationships with customers, physicians, and third-party payors are and will continue to be subject, directly and indirectly, to federal and state healthcare fraud and abuse laws and regulations. These laws also apply to the physicians and third-party payors who will play a primary role in the recommendation and prescription of our product candidates, if they become commercially available products. These laws may constrain the business or financial arrangements and relationships through which we might market, sell and distribute our products and will impact, among other things, any proposed sales, marketing and educational programs. There are also laws, regulations and requirements applicable to the award and performance of federal grants and contracts. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to them, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, disgorgement, the reimbursement of overpayments, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs, imprisonment, contractual damages, reputational harm, and diminished profits and earnings-any of which could adversely affect our ability to operate our business and our financial results.
Restrictions under applicable federal and state healthcare related laws and regulations include but are not limited to the following:
| ● | the federal Anti-Kickback Statute; |
| ● | the civil federal False Claims Act; |
| ● | the criminal federal False Claims Act; |
| ● | the Health Insurance Portability and Accountability Act, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations (collectively, “HIPAA”); |
| ● | the civil monetary penalties statute; |
| ● | federal transparency laws, including the federal Physician Sunshine Act (“PSA”); and |
| ● | analogous or similar state, federal, and foreign laws, regulations, and requirements. |
14
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations involve substantial costs. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other laws, regulations, or other requirements that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, restitution exclusion from government funded healthcare programs, corporate integrity agreements, deferred prosecution agreements, debarment from government contracts and grants and refusal of future orders under existing contracts, contractual damages, the curtailment or restructuring of our operations and other consequences. If any of the physicians or other healthcare providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, that person or entity may be subject to criminal, civil, or administrative sanctions, including exclusions from government funded healthcare programs. Moreover, availability of any federal grant funds which we may receive or for which we may apply is subject to federal appropriations law. Such grant funding may also be withdrawn or denied due to a violation of the above laws and/or for other reasons.
Coverage and Reimbursement; Healthcare Reform
Sales of pharmaceutical products depend significantly on the extent to which coverage and adequate reimbursement are provided by third-party payers. Third-party payers include state and federal government health care programs, managed care providers, private health insurers, and other organizations. Although we currently believe that third-party payers will provide coverage and reimbursement for our product candidates, if approved, we cannot be certain of this. Third-party payers are increasingly challenging the price, examining the cost-effectiveness and reducing reimbursement for medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. The United States government, state legislatures, and foreign governments have continued implementing healthcare reform and cost containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. We might need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of our product candidates. Third-party payers might not consider the product candidates that we develop to be cost-effective and not cover or sufficiently reimburse for their use. It is time-consuming and expensive for us to seek coverage and reimbursement from third-party payers, as each payer will make its own determination as to whether to cover a product and at what level of reimbursement. Thus, one payer’s decision to provide coverage and adequate reimbursement for a product does not assure that another payer will provide coverage or that the reimbursement levels will be adequate. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates to the extent we choose to develop or sell any product candidates outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement, and privacy, can vary greatly from country to country.
Employees
As of December 31, 2025, we had four employees, all of whom are located in the United States. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Property
The Company owns no real property. We maintain a month-to-month membership providing mail services and shared space for meetings and other business activities at 3540 Toringdon Way, Suite 200, #250, Charlotte, North Carolina. The current rent is approximately $100 per month.
15
Litigation
From time to time, we may become involved in various lawsuits and legal proceedings that arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm business.
We are currently not aware of any pending legal proceedings to which we are a party or of which any of our property is the subject, nor are we aware of any such proceedings that are contemplated by any governmental authority.
Available Information
Our principal office is located at 3540 Toringdon Way, Suite 200, #250, Charlotte, NC 28277 and our telephone number is (888) 609-1498. Our website is www.adaptinbio.com, and we can be contacted at info@adaptinbio.com. We are subject to the informational requirements of the Exchange Act and file or furnish reports and other information with the SEC. Such reports and other information filed by us with the SEC will be available free of charge on our website at www.adaptinbio.com when such reports are available on the SEC’s website. The SEC maintains a website that contains reports, information statements and other information that issuers file electronically with the SEC at www.sec.gov.
The contents of the websites referred to above are not incorporated into this filing. Further, our references to the URLs for these websites are intended to be inactive textual references only.
ITEM 1A. RISK FACTORS.
As a smaller reporting company as defined in Item 10 of Regulation S-K (17 CFR § 229.10(f)(1), we are not required to include risk factors in this Report.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
ITEM 1C. CYBERSECURITY.
Our Chief Financial Officer (“CFO”), with assistance from our third-party managed services team, identifies, assesses and manages the Company’s cybersecurity threats and risks. The Company leverages its
Our cybersecurity program leverages the cybersecurity strategies of our third-party service provider to focus on all areas of our business, including cloud-based environments, networks, applications, disaster recovery, business continuity and controls and safeguards enabled through business processes and tools. Our systems are continuously monitored for threats and unauthorized access.
ITEM 2. PROPERTIES.
Our principal executive office is located in North Carolina under a month-to-month lease arrangement. We currently lease any office space that we use in our operations, and we do not own any real property. We believe that our facility space adequately meets our needs and that we will be able to obtain any additional operating space that may be required on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS.
We are currently not aware of any pending legal proceedings to which we are a party or of which any of our property is the subject, nor are we aware of any such proceedings that are contemplated by any governmental authority.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
16
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our Common Stock is not listed on a national securities exchange, an over-the-counter market or any other exchange. Therefore, there is no trading market, active or otherwise, for our Common Stock and our Common Stock may never be included for trading on any stock exchange, automated quotation system or any over-the-counter market.
Holders
As of March 26, 2026, there were 152 holders of our 8,786,229 shares of our Common Stock.
Dividends
The Company has not paid any cash dividends on its shares of its Common Stock to date. The payment of cash dividends in the future will be dependent upon our revenues and earnings, if any, capital requirements and general financial condition. The payment of any dividends will be within the discretion of the Board of Directors.
Performance Graph
Not applicable.
Recent Sales of Unregistered Securities
For information regarding the Company’s sales of unregistered securities during the fiscal year ended December 31, 2025, please refer to the following previously filed reports:
| ● | our Current Report on Form 8-K, filed on February 18, 2025; |
| ● | our Quarterly Report on Form 10-Q, filed on May 15, 2025; |
| ● | our Quarterly Report on Form 10-Q, filed on August 14, 2025; and |
| ● | our Current Report on Form 8-K, filed on December 23, 2025. |
ITEM 6. [RESERVED]
17
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion and analysis is attributable to the operations of Adaptin Bio, Inc. for the years ended December 31, 2025 and 2024. This discussion and analysis should be read in conjunction with our consolidated financial statements for the years ended December 31, 2025 and 2024 and the related notes thereto, which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Overview of our Business
We are a biopharmaceutical company pioneering a transformational approach to enhancing the transfer of therapeutics into the brain, facilitating the treatment of brain cancers and other unmet medical conditions. Our precision medicine technology, originally developed by researchers in the Department of Neurosurgery at Duke University, harnesses the human immune system’s ability to target, recognize, destroy or deliver therapeutics to specific cells, including cancer cells. Our mission is to be the global leader and pioneer of this new treatment paradigm, integrating recombinant technology, gene therapy and cell therapy to address the challenges of targeting and delivering effective therapies, including to the brain for cancer and other CNS indications.
We are closely working with the researchers at Duke University to translate preclinical proof of concept data of our proprietary platform technology, the BRiTE Platform, into human clinical trials. BRiTE is a translatable method to specifically target malignant glioma using a tumor-specific, fully human bispecific antibody that is designed to redirect the patients’ own T cells to recognize and destroy tumor cells. Our first application of BRiTE is APTN-101, a proprietary EGFRvIII x CD3 bispecific T cell engager may have the ability to eliminate malignant glioma tumors in a variety of aggressive preclinical orthotopic tumor models. We designed APTN-101 to specifically redirect T cells against tumors expressing a well-characterized, mutated form of EGFR on a number of tumor types, including glioblastoma, breast and lung cancer. APTN-101 has been recently accepted under an investigator-led IND to begin first-in-human studies in brain cancer. Our goal is to complete preclinical studies on additional product candidates and file multiple INDs.
Duke University Exclusive Licensing Agreement
Effective January 11, 2023, we entered into a patent license agreement (the “Duke License”) with Duke University, whereby Duke University granted us an exclusive license with a right to sublicense the precision medicine technology, which we intend to develop using our BRiTE Platform. As part of the consideration for the license, we issued Duke 75 shares of our common stock (that were then valued at $175.86 per share, or $13,189, representing 5% of our then issued and outstanding common stock on a fully diluted basis). As a result of the Merger and recapitalization and additional issuances of common stock, Duke University now holds 161,960 shares of our Common Stock, or approximately 1.2% of the outstanding shares of the Company on a fully diluted basis. We also agreed to make milestone payments and pay royalties to Duke University, as well as to reimburse Duke University for prior patent expenses, as set forth in more detail below.
Operations Overview
Since inception, we have devoted substantially all of our resources to supporting our product development efforts, raising capital to support and expand such activities, and providing general and administrative support for these operations. We operate our business using a significant outsourcing model. As such, our team is composed of a small group of employees who direct a significantly large number of team members, including vendors and consultants, to enable execution of our operational plans. We do not currently have any products approved for sale, and we will continue to incur significant research and development and general administrative expenses related to our operations.
Since inception, we have incurred significant operating losses. For the year ended December 31, 2025, we recorded a net loss of $5,167,569. As of December 31, 2025, we had an accumulated deficit of $9,291,801. We expect to continue to incur significant losses for the foreseeable future. We anticipate that a substantial portion of our capital resources and efforts in the foreseeable future will be focused on completing the necessary development activities required for applying for and obtaining regulatory approval for our product candidates and, subsequently, preparing for potential commercialization of our product candidates. As of December 31, 2025 and December 31, 2024, we had $459,174 and $34,085 in cash and cash equivalents, respectively.
18
We expect to continue to incur significant expenses and operating losses for at least the next several years. Our net losses may fluctuate significantly from period to period, depending on the timing of our planned clinical trials and expenditures on other research and development activities. We expect our expenses will increase substantially over time as we:
| ● | continue our ongoing and planned development of APTN-101, including pre-clinical activity and our Phase 1 investigator-led trial for the treatment of GBM; |
| ● | build a portfolio of product candidates through development, or the acquisition or in-license of drugs, product candidates or technologies; |
| ● | initiate additional preclinical studies and clinical trials for APTN-101 for any additional indications we may pursue and for any additional product candidates that we may pursue in the future; |
| ● | hire clinical, regulatory and scientific personnel; |
| ● | add operational, financial and management information systems and personnel, including personnel to support our product development efforts; and |
| ● | incur additional legal, accounting, insurance and other expenses associated with operating as a public company. |
The Macroeconomic Climate
The recent economic trends and political changes, including the rapidly changing tariff structure, may materially adversely affect our business and corresponding financial position and cash flows. While inflationary factors have trended down and interest rates are beginning to trend down, they still may impact our overhead costs and may adversely affect our operating results. While interest rates have recently been trending down, they remain high and present a challenge impacting the United States and global economies. Recent volatility in the major stock indices could also present challenges in accessing additional capital. Such factors could make it more difficult for us to obtain traditional financing on acceptable terms, if at all, in the future. Although we do not believe that inflation has had a material impact on our financial position or results of operations to date, we may experience increases in the near future on our operating costs, including our labor, due to supply chain constraints, consequences associated with pandemics or public health situations, the Russia-Ukraine war, and other U.S. geopolitical issues, such as recent U.S. military actions in Venezuela and Iran and the ongoing implementation of new tariff structures and subsequent changes thereto, affecting other territories and employee availability and wage increases, all of which may result in additional stress on our working capital resources.
Components of Results of Operations
Research and Development Expenses
Research and development expenses consist primarily of fees paid to third-party service providers and, in 2025, personnel costs and other personnel-related compensation expenses, including stock-based compensation costs. Research and development costs are expensed in the periods in which they are incurred. Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors, collaborators and third-party service providers.
To date, substantially all our research and development expenses have been related to the licensing and preclinical development of APTN-101. As we progress, we expect our research and development costs to increase for additional preclinical and clinical development of APTN-101 in GBM.
19
The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming and is subject to uncertainties and delays. As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of our product candidates, if at all.
General and Administrative Expenses
General and administrative expenses include expenses for executive compensation and related costs, stock-based compensation, outside professional services and other general administrative expenses, including costs associated with the Merger. Outside professional services consist of patent maintenance expenses, legal, accounting, insurance and audit services and other consulting fees.
We also expect to continue to incur expenses as a public company, including expenses related to compliance with SEC rules and regulations and those of any national securities exchange on which our securities are traded, additional insurance expenses, investor relations activities, and other administrative and professional services.
Interest Expense
Interest expense primarily consists of contractual debt interest expense, the amortization of debt issuance costs and the amortization of discounts arising from bifurcated derivative liabilities, prior to extinguishment.
Results of Operations
Comparison of the Years Ended December 31, 2025 and 2024
The following table summarizes our results of operations and changes for the periods indicated:
| For the Years Ended December 31, | ||||||||||||||||
| 2025 | 2024 | $ Change | % Change | |||||||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | $ | 1,774,166 | $ | 1,824,591 | $ | (50,425 | ) | -3 | % | |||||||
| General and administrative | 3,640,777 | 745,294 | 2,895,483 | 389 | % | |||||||||||
| Total Operating Expenses | 5,414,943 | 2,569,885 | 2,845,058 | 111 | % | |||||||||||
| Loss from Operations | (5,414,943 | ) | (2,569,885 | ) | (2,845,058 | ) | 111 | % | ||||||||
| Other Expense: | ||||||||||||||||
| Interest Expense | 72,659 | 341,048 | (268,389 | ) | -79 | % | ||||||||||
| Loss on fair value of derivative liability | 6,312 | 17,087 | (10,775 | ) | -63 | % | ||||||||||
| (Gain) loss on extinguishment of debt | (326,345 | ) | 194,537 | (520,882 | ) | -268 | % | |||||||||
| Total Other Expenses | (247,374 | ) | 552,672 | |||||||||||||
| Net Loss | $ | (5,167,569 | ) | $ | (3,122,557 | ) | $ | (2,045,012 | ) | 65 | % | |||||
20
Research and Development Expenses
Research and development expenses decreased by $50,425, or 3%, for the year ended December 31, 2025 when compared to research and development expenses for the year ended December 31, 2024. During 2025, the decrease in research and development expenses is primarily attributable to the completion of our repeat-dose toxicology study of APTN-101 that began in late 2023 along with costs related to our assay development program as it nears completion that resulted in a decrease of approximately $1.5 million. Offsetting those decreases were increased costs related to our sponsored research agreements with Duke University of approximately $776,000, an increase in clinical trial packaging costs of approximately $198,000, the minimum annual royalty for 2025 of $25,000, increases in post-Merger compensation and related costs for research and development personnel of $95,000 and the stock-based compensation expense for stock options granted during the period of approximately $117,000. Costs incurred during the year ended December 31, 2024 consist primarily of costs for the ongoing assay development and repeat-dose toxicology studies with our third-party service providers.
General and Administrative Expenses
General and administrative expenses increased by approximately $2.9 million, or 389%, for the year ended December 31, 2025 compared to the year ended December 31, 2024. The increase was primarily related to increases in legal, accounting and consulting costs related to the Merger and filings with the SEC of approximately $665,000, increases in post-Merger compensation and related costs for our executive officers of approximately $960,000, stock-based compensation costs recorded in conjunction with options granted during the period of approximately $380,000 and increases in our directors fees, stock quotation fees, insurance costs, financial printing and transfer agent costs as a public company of approximately $500,000 in the aggregate. Additionally, we recorded $250,000 of expense related to the issuance of stock to a third-party vendor. Costs incurred during the year ended December 31, 2024 consisted primarily of legal fees, accounting fees and consulting fees.
Interest Expense
Interest expense decreased $268,389, or 79%, for the year ended December 31, 2025 when compared to the year ended December 31, 2024. The decrease in interest expense was related to the conversion of all outstanding debt in conjunction with the Merger and Offering in February 2025. For the year ended December 31, 2024, we recorded interest expense, debt issuance costs amortization, discounts related to derivative liability amortization and loss on derivative liabilities for our then outstanding debt.
Other Income and Expense
Prior to the Merger, we had recorded the accrued interest, debt issuance costs amortization, discounts related to derivative liability amortization and related costs of the 2024 Bridge Notes. Upon completion of the issuance of the 2024 Bridge Notes and based on the information then currently available, the recorded bifurcated derivative liability related to the embedded redemption feature of this debt was $333,333. In December 2024, we also recorded a bifurcated derivative liability related to the Exchange Notes executed by holders of the 2023 Bridge Notes of $194,537, that due to the deemed extinguishment of the 2023 Bridge Notes upon execution of the Exchange Notes, gave rise to a loss on extinguishment of $194,537 for the year ended December 31, 2024. At the date of the Merger, the carrying value of the derivative liability totaled $551,269. At the Initial Closing of the Offering, the $1,500,000 aggregate principal amount of Exchange Notes and 2024 Bridge Notes, plus accrued interest thereon, automatically converted into shares of our common stock. As a result of the conversion, we recorded a gain on debt extinguishment of $326,345.
Liquidity and Capital Resources
Bridge Financings
We raised bridge financing through the offer and sale (a) in 2023 of $500,000 principal amount of our 10% Secured Promissory Notes (the “2023 Bridge Notes”) (including warrants to purchase up to 56,815 shares of our Common Stock at an exercise price of $4.40 per share) and (b) in 2024 of $1,000,000 principal amount of its 10% Secured Subordinated Convertible Promissory Notes (the “2024 Bridge Notes”), which in each case were sold to a limited number of accredited investors pursuant to Regulation D under the Securities Act. In December 2024, the 2023 Bridge Notes were cancelled and exchanged for $500,000 principal amount of our 10% Secured Convertible Promissory Notes (the “Exchange Notes”). In connection with the note exchange, the holders of the 2023 Bridge Notes were also issued warrants to purchase up to 75,755 shares of our Common Stock at an exercise price of $3.30 per share. The Exchange Notes, collectively with the 2024 Bridge Notes, are referred to herein as the “Bridge Notes”.
21
Pre-Merger Warrants
As described above, prior to the Merger, we raised bridge financing through the offer and sale of the 2023 Bridge Notes. We agreed to issue common stock warrants (the “2023 Bridge Note Warrants”) to the purchasers of the 2023 Bridge Notes. The 2023 Bridge Note Warrants give the holders the right to purchase an aggregate of up to 56,815 shares of our Common Stock at an exercise price of $4.40 per share. In December 2024, the 2023 Bridge Notes were cancelled and exchanged for the Exchange Notes. In connection with this note exchange, the holders of the 2023 Bridge Notes were issued warrants (the “Exchange Warrants”) to purchase an aggregate of up to 75,755 shares of our Common Stock at an exercise price of $3.30 per share. The 2023 Bridge Note Warrants and the Exchange Warrants must be exercised on or prior to the close of business on February 11, 2030, which is the fifth anniversary of the initial closing of the Private Placement. We refer to the 2023 Bridge Note Warrants and the Exchange Warrants collectively as the “Pre-Merger Warrants.”
Private Placements
Concurrent with the closing of the Merger, we sold, in an initial closing (the “Initial Closing”) of a private placement offering (the “Offering”), 1,080,814 units (the “Units”) at a purchase price of $4.40 per Unit, each consisting of (i) one share of common stock, (ii) a one-year warrant to purchase one share of our Common Stock at an exercise price of $4.40 per share, and (iii) a five-year warrant to purchase one-half of a share of our Common Stock at an exercise price of $6.60 per share. On March 31, 2025, we sold in the final closing of our Offering, 319,529 Units for an aggregate purchase price of $1,405,923.
At the Initial Closing of the Offering in February 2025, the $1,500,000 aggregate principal amount of outstanding Bridge Notes, plus accrued interest thereon, converted automatically into shares of our Common Stock at a conversion price of $3.30 per share, or 501,140 shares of common stock in the aggregate, and the holders of the 2023 Bridge Notes were issued, pursuant to existing agreements, warrants to purchase up to 132,570 shares of our Common Stock at an exercise price of $3.30 or $4.40 per share and with a term of five years. Further, as set forth above, we raised gross proceeds in our Offering of $6,161,505 through the issuance of Units. Additionally, in December 2025, we raised gross proceeds of $1,000,000 through the sale of 200,000 shares of our common stock. In the aggregate, after factoring in offering costs, we recorded net proceeds in 2025 of $5,284,105.
In December 2025, we sold, in an initial closing of a private placement offering, 200,000 shares of our common stock at an aggregate purchase price of $1,000,000, or $5.00 per share. The offering period commenced on October 29, 2025 and was scheduled to continue until the later of (i) January 31, 2026, unless extended by the Company and the placement agent; (ii) the date on which the maximum offering amount of approximately $4.0 million (the “Maximum Offering”) was sold by the Company; or (iii) on a date mutually agreed upon in writing by the Company and the placement agent (the “Offering Period”). On December 30, 2025, the Company and the placement agent agreed to extend the offering period to February 27, 2026; on February 28, 2026 was extended until March 31, 2026, and; on March 31, 2026 was extended until April 30, 2026. As of the date of the Initial Closing, the Company recorded net proceeds of $582,961, net of costs of the transaction of $417,039 that had been incurred as of that date.
Accordingly, as of December 31, 2025, the Company had total assets equal to $619,159 comprised of cash and prepaid assets. The Company’s current liabilities as of December 31, 2025, totaled $2,108,555, and were comprised of accounts payable and accrued liabilities. Accordingly, as of December 31, 2025, we had cash and cash equivalents, working capital deficit and accumulated deficit of $459,174, $1,489,396 and $9,291,801, respectively.
Based on our current operating plan, we anticipate that our existing cash balance will not be sufficient to fund our operating activities for the next twelve months and, as such, substantial doubt exists about our ability to support our operations and fund our obligations for next twelve months from the date of issuance of these consolidated financial statements. We plan to continue to fund our losses from operations through cash on hand, as well as through future equity offerings and debt financings, or other third-party funding. There can be no assurance that additional funds will be available when needed from any source or, if available, will be available on terms that are acceptable to us. Even if we raise additional capital, we may also be required to modify, delay or abandon some of our plans which could have a material adverse effect on our business, operating results and financial condition and our ability to achieve our intended business objectives. Any of these actions could materially harm our business, results of operations and future prospects.
22
Cash Flows
The following is a summary of the Company’s cash flows provided by (used in) operating and financing activities during the years ended December 31, 2025 and 2024:
| For the Years Ended December 31, | ||||||||||||||||
| 2025 | 2024 | $ Change | % Change | |||||||||||||
| Net cash (used in) provided by: | ||||||||||||||||
| Operating activities | (4,777,892 | ) | (649,283 | ) | (4,128,609 | ) | -636 | % | ||||||||
| Financing activities | 5,202,981 | 678,614 | 4,524,367 | 667 | % | |||||||||||
| Net increase in cash during the periods | 425,089 | 29,331 | 395,758 | 1349 | % | |||||||||||
Net Cash Used in Operating Activities
For the years ended December 31, 2025 and 2024, we used cash for operating activities of $4,777,892 and $649,283, respectively. Our cash use for the year ended December 31, 2025 was primarily attributable to our net loss of $5,167,569, adjusted for net non-cash expenses in the aggregate amount of $480,553, less $90,876 of net cash used in changes in operating assets and liabilities. Our cash use for the year ended December 31, 2024 was primarily attributable to our net loss of $3,122,557, adjusted for net non-cash expenses in the aggregate amount of $451,413, and offset by $2,021,861 of cash provided by changes in of operating assets and liabilities.
Net Cash Provided by Financing Activities
During the year ended December 31, 2025, cash provided by financing activities was $5,202,981, of which, $7,161,504 was provided by offering proceeds related to the offerings, offset by $1,683,523 of payments of equity issuance costs and $275,000 of repayment of notes payable to a related party. During the year ended December 31, 2024, cash provided by financing activities was $678,614, of which $872,490 was related to net proceeds from the issuance of notes payable offset by $193,876 of equity issuance costs paid in advance of the February 2025 Offering.
Funding Requirements
We use our cash primarily to fund research and development expenditures and the administrative costs of operating a public company. We expect our research and development expenses to increase in 2026 as we move forward with our Phase 1 program in GBM for APTN-101 and incur additional pre-clinical expenses for any additional programs we may elect to pursue. We expect to incur a decrease in general and administrative expenses in 2026 primarily related the costs of the Merger and Offering in 2025 not recurring offset by continued support of our increasing research and development activities and costs associated with being a publicly held company, including professional fees, personnel, insurance and regulatory compliance related costs. We expect to incur operating losses for the foreseeable future as we continue the preclinical and clinical development of our product candidate. At this time, due to the inherently unpredictable nature of clinical development, we cannot reasonably estimate the costs we will incur and the timelines that will be required to complete development, obtain marketing approval, and commercialize APTN-101 or any future product candidates, if at all. For the same reasons, we are also unable to predict when, if ever, we will generate revenue from product sales or whether, or when, if ever, we may achieve profitability. Clinical and preclinical development timelines, the probability of success, and development costs can differ materially from expectations.
The timing and amount of our operating expenditures will depend largely on:
| ● | the timing, progress and results of our ongoing and planned preclinical and clinical development activities for APTN-101 in GBM; |
| ● | the scope, progress, results and costs of preclinical development, testing and clinical trials of APTN-101 for any additional indications; |
| ● | the ability of our vendors and third-party service providers to accurately forecast expenses and deliver on expectations; |
23
| ● | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; and |
| ● | the extent to which we acquire or in-license other product candidates and technologies. |
Until such time, if ever, as we can generate substantial revenue from product sales, we expect to fund our operations and capital funding needs through equity and/or debt financing. We may also consider entering into collaboration arrangements or selectively partnering for clinical development and commercialization. The sale of additional equity would result in additional dilution to our shareholders. The incurrence of debt financing would result in debt service obligations and the instruments governing such debt could provide for operating and financing covenants that restrict our operations or our ability to incur additional indebtedness, among other items. If we are not able to secure adequate additional funding, we may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, and/or suspend or curtail planned programs. Any of these actions could materially and adversely affect our business, financial condition and results of operations.
Contractual Obligations and Commitments
Duke License
In January 2023, we entered into the Duke License for an exclusive, world-wide, sub-licensable license to precision medicine technology. As a component of the Duke License, we agreed to make payments based on clinical and commercial milestones and continuing royalty payments on any sales made after approval by regulatory authorities. These milestones include initiation of Phase II or Phase III clinical trials, submission of applications for market approval in multiple jurisdictions including the United States, European Union and Japan and the initiation of post-approval commercial sales in the same jurisdictions. Based on an assumption that all milestones related to the current development program are met during the course of the Duke License, these milestone payments would total approximately $11.7 million. As of December 31, 2025, we had not met any milestones as defined in the agreement and, accordingly, have recorded no expense or liability related to such payments.
We also agreed to pay royalties equal to low- to mid- single digit percentages of annual net sales on a country-by-country and product-by-product basis subject to downward adjustment to low single digit percentages of our net annual sales in the event there is no valid claim of a patent for the product, with minimum annual royalty levels established. We also must pay Duke percentages in the low tens, twenties and thirties of sublicensing fees after initiation of the first Phase III study, after initiation of the first Phase II study but prior to initiation of the first Phase III study, and prior to initiation of the first Phase II study, respectively, as set forth in the Duke License. Based on the minimum annual royalty levels established in the Duke License, we accrued $25,000 for royalties due under the agreement as of December 31, 2025.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet financing arrangements or any relationships with unconsolidated entities or financial partnerships, such as structured finance or special purpose entities, that were established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Critical Accounting Estimates
The preparation of financial statements and related disclosures in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and income and expenses during the periods reported. We base our estimates on our limited historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
24
We consider an accounting estimate to be critical if: (i) the accounting estimate requires us to make assumptions about matters that were highly uncertain at the time the accounting estimate was made, and (ii) changes in the estimate that are reasonably likely to occur from period to period or use of different estimates that we reasonably could have used in the current period, would have a material impact on our financial condition or results of operations.
Significant estimates and assumptions reflected in the consolidated financial statements relate to and include, but are not limited to, the fair value of warrants, the fair value of stock issued in exchange for services, prepaid expenses and accrued liabilities that are measured based on progress toward completion of research and development projects, and the grant date fair value of stock options granted to employees, consultants and directors, and the resulting stock-based compensation expense, calculated using the Black-Scholes option-pricing model.
Future events and their effects cannot be predicted with certainty; accordingly, accounting estimates require the exercise of judgment. Accounting estimates used in the preparation of these consolidated financial statements change as new events occur, as more experience is acquired, as additional information is obtained and as the operating environment changes.
We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates:
a) Research & Development Expenses — Accruals
Research and development costs are charged to expense in the period of expenditure. Our research and development costs consist primarily of compensation and related costs and fees paid to external, third-party service providers, such as Contract Research Organizations (“CRO”s) or Contract Manufacturing Organizations (“CMO”s).
Research and development expenses include direct costs associated with CROs for research activities and direct CMO costs for the manufacture, formulation and packaging of clinical trial material. Direct costs associated with our CROs and CMOs are generally payable on a time-and-materials basis, or when milestones are achieved. The invoicing from clinical trial sites can lag several months or can be in advance of the work to be completed. We record expenses for our clinical trial activities performed by third parties based upon estimates of the percentage of work completed of the total work over the life of the individual trial in accordance with agreements established with CMOs and CROs. We determine the estimates through discussions with internal clinical personnel, CROs and CMOs as to the progress or stage of completion of trials or services and the agreed-upon fee to be paid for such services based on facts and circumstances known to us as of each consolidated balance sheet date. The actual costs and timing of clinical trials are highly uncertain, subject to risks and may change depending upon a number of factors, including our clinical development plan. If the actual timing of the performance of services of the level of effort varies from the estimate, we will adjust the accrual accordingly.
b) Stock-Based Compensation
We recognize compensation costs related to share options granted to employees, consultants and directors based on the estimated fair value of the awards on the date of grant. We estimate the grant date fair value, and the resulting stock-based compensation expense, using the Black Scholes option pricing model. This Black Scholes option pricing model uses various inputs to measure fair value, including estimated fair value of our underlying common shares at the grant date, expected term, estimated volatility, risk-free interest rate and expected dividend yields of our common shares. The estimated volatility creates a critical estimate because we have not been a public company long enough and do not have sufficient trading history to demonstrate our own historical volatility. As such, we estimate volatility using an internally developed peer group of similar companies. The grant date fair value of the stock-based awards is recognized on a straight-line basis over the requisite service periods, which are generally the vesting period of the respective awards or based upon the terms of performance obligations, if any. Forfeitures are accounted for as they occur.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information otherwise required under this item.
25
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
Financial Statements:
Index to Consolidated Financial Statements:
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Adaptin Bio, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Adaptin Bio, Inc. as of December 31, 2025 and 2024, and the related consolidated statements of operations, changes in stockholders’ deficit, and cash flows for each of the two years in the period ended December 31, 2025, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2025 and 2024, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2025, in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt Regarding Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the entity’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the entity’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
We have served as Adaptin Bio, Inc’s auditor since 2023.
April 1, 2026
PCAOB ID Number
F-2
ADAPTIN BIO, INC.
CONSOLIDATED BALANCE SHEETS
| December 31, | December 31, | |||||||
| 2025 | 2024 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Prepaid expenses and other current assets | ||||||||
| Total Current Assets | ||||||||
| Non-Current Assets: | ||||||||
| Deferred equity issuance costs | ||||||||
| Total Assets | $ | $ | ||||||
| Liabilities and Stockholders’ Deficit | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable- trade | $ | $ | ||||||
| Accrued expenses | ||||||||
| Convertible notes payable, net of debt issuance costs and discounts | ||||||||
| Derivative liability arising from convertible notes payable | ||||||||
| Accrued interest | ||||||||
| Total Current Liabilities | ||||||||
| Total Liabilities | ||||||||
| Commitments and contingencies (Note 11) | ||||||||
| Stockholders’ Deficit: | ||||||||
| Preferred stock, $ December 31, 2025 and 2024 | ||||||||
| Common stock, $ | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Deficit | ( | ) | ( | ) | ||||
| Total Liabilities and Stockholders’ Deficit | $ | $ | ||||||
| [1] |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
ADAPTIN BIO, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| For the Year Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Operating Expenses: | ||||||||
| Research and development | $ | $ | ||||||
| General and administrative | ||||||||
| Total Operating Expenses | ||||||||
| Loss from Operations | ( | ) | ( | ) | ||||
| Other Expense (Income): | ||||||||
| Interest expense | ||||||||
| Loss on change in fair value of derivative liabilities | ||||||||
| (Gain) loss on extinguishment of debt | ( | ) | ||||||
| Total Other Expense (Income), net | ( | ) | ||||||
| Loss Before Provision for Income Taxes | ( | ) | ( | ) | ||||
| Provision for income taxes | ||||||||
| Net Loss | $ | ( | ) | $ | ( | ) | ||
| Net Loss Per Share | ||||||||
| Basic and diluted [1] | $ | ( | ) | $ | ( | ) | ||
| Weighted Average Common Shares Outstanding: | ||||||||
| Basic and diluted[1] | ||||||||
| [1] |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
ADAPTIN BIO, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ DEFICIT
| For the Year Ended December 31, 2025 | ||||||||||||||||||||
| Additional | Total | |||||||||||||||||||
| Common Stock [1] | Paid-In | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balance, December 31, 2024 – Prior to Recapitalization | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||
| Recapitalization | ( | ) | ||||||||||||||||||
| Balance, December 31, 2024 – Following the Recapitalization | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||
| Equity of Unite Acquisition 1 Corp. at the time of the exchange | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Common stock cancelled at the time of the exchange | ( | ) | ( | ) | ||||||||||||||||
| Recapitalization of Unite Acquisition 1 Corp. accumulated deficit at time of the exchange | - | ( | ) | |||||||||||||||||
| Common stock and warrants issued in private placements [2] | ||||||||||||||||||||
| Common stock and warrants issued in connection with debt extinguishment | ||||||||||||||||||||
| Forgiveness of accrued consulting fees by related parties | - | |||||||||||||||||||
| Shares issued in exchange for services | ||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net Loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2025 | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||
| [1] |
| [2] |
The accompanying notes are an integral part of these consolidated financial statements.
F-5
ADAPTIN BIO, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ DEFICIT
| For the Year Ended December 31, 2024 | ||||||||||||||||||||
| Additional | Total | |||||||||||||||||||
| Common Stock [1] | Paid-In | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balance, January 1, 2024 | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2024 | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||
| [1] | The number of shares and per share value of the Company’s common stock have been retroactively recast to reflect the exchange ratio pursuant to the Merger (see Note 3). |
The accompanying notes are an integral part of these consolidated financial statements.
F-6
ADAPTIN BIO, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(all amounts in USD)
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization of debt issuance costs and discounts | ||||||||
| Change in fair value of derivative liabilities | ||||||||
| Loss (Gain) on extinguishment of debt | ( | ) | ||||||
| Shares issued in exchange for services | ||||||||
| Stock-based compensation | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | ( | ) | ( | ) | ||||
| Accounts payable- trade | ||||||||
| Accrued expenses | ( | ) | ||||||
| Accrued interest | ||||||||
| Net Cash Used In Operating Activities | ( | ) | ( | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of convertible notes payable | ||||||||
| Repayment of notes payable- related party | ( | ) | ||||||
| Proceeds from issuance of common stock and warrants in private placements | ||||||||
| Payment of issuance costs related to private placements | ( | ) | ( | ) | ||||
| Payment of debt issuance costs | ( | ) | ||||||
| Net Cash Provided By Financing Activities | ||||||||
| Net Increase In Cash and Cash Equivalents | ||||||||
| Cash and Cash Equivalents - Beginning of Year | ||||||||
| Cash and Cash Equivalents - End of Year | $ | $ | ||||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Cash paid for: | ||||||||
| Interest | $ | $ | ||||||
| Income taxes | $ | $ | ||||||
| Non-cash investing and financing activities: | ||||||||
| Recapitalization of Unite Acquisition 1 Corp. accumulated deficit at time of the exchange | $ | ( | ) | $ | ||||
| Common stock cancelled at the time of the exchange | $ | $ | ||||||
| Convertible notes and accrued interest converted into common stock | $ | $ | ||||||
| Forgiveness of accrued consulting fees by related parties | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
ADAPTIN BIO, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. BUSINESS ORGANIZATION, NATURE OF OPERATIONS, BASIS OF PRESENTATION, AND RISKS AND UNCERTAINTIES
Organization and Operations
On February 11, 2025, Adaptin Bio, Inc. completed the business combination contemplated by that certain Agreement and Plan of Merger and Reorganization, dated as of February 11, 2025, by and among Unite Acquisition 1 Corp. (“Unite Acquisition”), a public shell company incorporated in the state of Delaware on March 10, 2022, its wholly-owned subsidiary, Adaptin Acquisition Co., a Delaware corporation formed in the State of Delaware on January 30, 2025 (“Merger Sub”), and Adaptin Bio Operating Corporation (formerly “Adaptin Bio, Inc.”) (the “Merger”). Upon the completion of the Merger, Unite Acquisition changed its name to “Adaptin Bio, Inc.”
Adaptin Bio, Inc. is dedicated to the development and commercialization of products utilizing novel technology that enhances the delivery of drugs and other compounds to the brain and other tissues for a variety of indications. The Company’s novel technology was originally developed by researchers in the Department of Neurosurgery at Duke University and licensed by the Company in 2023. The Company’s technology is engineered to facilitate the transport of therapeutics to tissues of interest, including the brain, potentially generating improved treatments for solid tumors and central nervous system (“CNS”) disorders.
The Merger was accounted for as a reverse recapitalization as Adaptin Bio Operating Corporation was determined to be the accounting acquirer under Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 805, Business Combinations. Please refer to Note 3 - Reverse Recapitalization for additional details of the Merger.
Upon the completion of the Merger, the share, per share value and net loss per share in the accompanying consolidated financial statements for all dates prior to the Merger were retroactively recast to reflect the exchange ratio pursuant to the Merger.
Basis of Presentation and Principles of Consolidation
The Company’s consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for financial information and include the accounts of the Company’s consolidated subsidiary. All intercompany balances and transactions have been eliminated in consolidation. Any reference in these notes to applicable guidance is meant to refer to the authoritative U.S. GAAP as found in the ASC and Accounting Standards Update (“ASU”) of the FASB.
Emerging Growth Company
The Company is an “emerging growth company” and has elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows the Company to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies.
Going Concern
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. As of December 31, 2025, the Company had cash and cash equivalents of $
F-8
Management has evaluated the Company’s operating plan against its existing cash and cash equivalents and determined that substantial doubt exists about the Company’s ability to support its operations and fund its obligations for the next twelve months from the date of issuance of these consolidated financial statements.
The Company plans to continue to fund its losses from operations through cash and cash equivalents on hand, as well as cash from future equity offerings, debt financings, or other third-party funding. There can be no assurance that additional funds will be available when needed or, if available, will be available on terms that are acceptable to the Company. Even if the Company raises additional capital, it may also be required to modify, delay or abandon some of its plans which could have a material adverse effect on its business, operating results and financial condition and its ability to achieve its intended business objectives. Any of these actions could materially harm the Company’s business, results of operations and future prospects. These financial statements have been prepared on a going concern basis and do not include any adjustments to the amounts and classification of assets and liabilities that may be necessary in the event the Company can no longer continue as a going concern.
Significant Risks and Uncertainties
The Company is subject to challenges and risks specific to its business and its ability to execute on its strategy, as well as risks and uncertainties common to companies in the pharmaceutical industry, including, without limitation, risks and uncertainties associated with obtaining regulatory approval of its product candidate; delays or problems in the supply of its study drug or failure to comply with manufacturing regulations; identifying, acquiring or in-licensing product candidates; pharmaceutical product development and the inherent uncertainty of clinical success; and the challenges of protecting and enhancing its intellectual property rights; and complying with applicable regulatory requirements.
Further, the Company may be impacted by general economic, political, and market conditions, including deteriorating market conditions due to investor concerns regarding inflation, armed conflicts, and overall fluctuations in the financial markets in the U.S. and abroad.
2. SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amount of expenses during the reporting period. Actual results could differ from those estimates.
Significant estimates and assumptions reflected in the consolidated financial statements relate to and include, but are not limited to, the fair value of derivative liabilities, the fair value of stock issued in exchange for services, accrued liabilities that are measured based on progress toward completion of research and development projects, and the grant date fair value of stock options granted to employees, consultants and directors, and the resulting stock-based compensation expense, calculated using the Black-Scholes option-pricing model.
Future events and their effects cannot be predicted with certainty; accordingly, accounting estimates require the exercise of judgment. Accounting estimates used in the preparation of these consolidated financial statements change as new events occur, as more experience is acquired, as additional information is obtained and as the operating environment changes.
F-9
Segments
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker (“CODM”) in making decisions regarding resource allocation and assessing performance. The Company views its operations and manages its business as a single segment, which seeks to develop and commercialize products by utilizing novel technology that enhances the delivery of drugs and other compounds to the brain and other tissues for a variety of indications. The accounting policies of the segment are the same as those described in the summary of significant accounting policies. The CODM, who is the Company’s chief executive officer, utilizes the Company’s financial information on an aggregate basis for purposes of making operating decisions, allocating resources and assessing financial performance, as well as for making strategic operations decisions and managing the organization. The CODM is not regularly provided with disaggregated actual expense information, other than the actual expense information included in the consolidated statements of operations. The measure of segment assets is reported on the consolidated balance sheets as total assets. The Company has not yet generated any revenue from product sales.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of 90 days or less on the date of purchase to be cash equivalents. The carrying value of cash and cash equivalents approximates fair value due to the short-term nature of these items.
Currently, all cash is deposited in two financial institutions. The balances are insured by the Federal Deposit Insurance Corporation (“FDIC”) up to certain limits. In the event there is cash in the banks, it may, at times, exceed FDIC insurable limits. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial position.
Prepaid Expenses
Prepaid expenses consist primarily of research and development costs paid in advance of the performance of activities, professional fee retainers and insurance premiums paid at policy inception.
Deferred Equity Issuance Costs
Deferred equity issuance costs represent amounts paid for legal, accounting, consulting, and other offering expenses in conjunction with an anticipated future raise of additional capital. These costs are netted against additional paid-in capital as a cost of the equity issuance upon closing of the respective equity placement.
As of December 31, 2024, there were $
Derivative Liability
The Company does not use derivative instruments to hedge exposures to cash flow, market, or foreign currency risks. The Company analyzes all financial instruments, including its convertible debt under the FASB ASC Topic No. 815, Derivatives and Hedging (“ASC 815”), to determine if such instruments contain features that qualify as embedded derivatives. The accounting treatment of derivative financial instruments requires that the Company record bifurcated embedded conversion features and any related freestanding instruments at their fair values as of the inception date of the agreement and at fair value as of each subsequent balance sheet date. Any increase or decrease in the fair value would be recorded in the results of operations within other income/expense as change in fair value of derivative liabilities. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is reassessed at the end of each reporting period.
Upon conversion or repayment of a debt or equity instrument in exchange for equity shares, where the embedded conversion feature has been bifurcated and accounted for as a derivative liability (generally convertible debt), the Company would record the equity shares at fair value on the date of conversion, relieve the carrying value of all related debt, derivative liabilities (after marking them to fair value), and unamortized debt discounts, and recognize a net gain or loss on debt extinguishment, if any.
F-10
Accrued Expense
Accrued expenses consist primarily of research and development, legal, accounting, consulting and license-related fees.
Warrants
The Company accounts for its warrants in accordance with the guidance in ASC 815-40-15. This guidance provides that if the warrants do not meet the criteria for equity treatment, the warrants must be recorded as an asset or a liability. The Company estimates the fair value of warrants using a Black-Scholes valuation model, which requires the use of multiple subjective inputs including estimated future volatility, risk free rate and the expected terms of the warrant.
Research and Development
Research and development expenses include direct costs associated with both pre-clinical and clinical activities and will, in future periods, include costs associated with the Company’s clinical trials including CRO fees, direct CMO costs for additional manufacture and packaging of trial material, as well as investigator and patient related costs at sites at which the Company’s clinical trials will be conducted. Direct costs associated with the Company’s CROs and CMOs are generally payable on a time and materials basis, or when milestones are achieved. The invoicing from third-party vendors can be for work performed on a time and material basis or reflecting milestones outlined in the initial contract for services and may not reflect actual work performed as of a specific measurement date. The Company records expenses for its pre-clinical and clinical activities performed by third parties based upon estimates of the percentage of work completed of the total work over the life of the individual study in accordance with agreements established with the third-party vendors. The Company determines the estimates through discussions with development personnel and its third-party vendors as to the progress or stage of completion of trials or services and the agreed upon fee to be paid for such services based on facts and circumstances known to the Company as of each balance sheet date. The actual costs and timing of clinical trials are highly uncertain, subject to risks and may change depending upon a number of factors, including the Company’s clinical development plan. If the actual timing of the performance of services of the level of effort varies from the estimate, the Company will adjust the accrual accordingly. If the receipt of invoices is in advance of estimated work performed, the Company will record a prepaid expense as of the measurement date.
Research and development expenses also include the cost of in-process research and development (“IPR&D”) assets purchased in an asset acquisition transaction. IPR&D assets are expensed unless the assets acquired are deemed to have an alternative future use, provided that the acquired assets did not also include processes or activities that would constitute a business as defined under U.S. GAAP, the technology has not received regulatory approval for marketing and, absent obtaining such approval, has no established alternative future use. Acquired IPR&D payments are immediately expensed in the period in which they are incurred and include upfront payments, as well as subsequent pre-commercial milestone payments. Research and development costs after the acquisition are expensed as incurred. The costs associated with maintaining the patents after the acquisition are the responsibility of the Company and will be expensed as incurred as a general and administrative expense.
Research and development costs are charged to expense as costs are incurred in performing research and development activities. To date, the Company’s research and development costs consist primarily of costs related to obtaining the license to its technology (see Note 11), conducting pre-clinical IND-enabling activities, the manufacture and packaging of clinical trial materials and start-up costs related to the Phase 1 clinical program that is anticipated to begin in 2026. It is anticipated that additional expenses in future periods for both pre-clinical and clinical activities will consist primarily of fees paid to contract research organizations (“CROs”) and to contract manufacturing organizations (“CMOs”).
Income Taxes
Deferred income taxes are provided on temporary differences between financial statement and income tax reporting. Temporary differences are differences between the amounts of assets and liabilities reported for financial statement and income tax purposes.
F-11
Deferred tax assets are recognized for temporary differences that will be deductible in future years’ tax returns and for operating loss and tax credit carryforwards. Deferred tax assets are recognized only if it is more likely than not that a tax position will be realized or sustained upon examination by the relevant taxing authority. A tax position that meets the more-likely-than-not recognition threshold is initially and subsequently measured as the largest amount of tax benefit that has a greater than
Deferred tax assets are reduced by a valuation allowance if it is deemed more likely than not that some or all of the deferred tax assets will not be realized. Deferred tax liabilities are recognized for temporary differences that will be taxable in future years.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740) - Improvements to Income Tax Disclosures, which enhanced income tax disclosures that reflect how operations and related tax risks, as well as how tax planning and operational opportunities, affect the tax rate and prospects for future cash flows. This standard was adopted by the Company for its annual reporting beginning January 1, 2025. The adoption of ASU 2023-09 has expanded the Company’s income tax disclosures for the year ended December 31, 2025 and future periods, but had no material impact on income tax benefit or expense or related tax assets or liabilities.
Net Loss per Share
Basic net loss per common share is calculated by dividing net loss by the weighted-average number of common shares outstanding for the period, without consideration for potentially dilutive securities. For the periods presented, basic and diluted net loss per common share are identical as the Company has incurred losses for both periods. For diluted net loss per share, the weighted-average number of shares of common stock is the same for basic net loss per share because when a net loss exists, potentially dilutive securities are not included in the calculation when the impact is anti-dilutive. The Company has potentially dilutive securities in the form of common stock warrants and stock options issued during the year ended December 31, 2025 and the number of shares available upon conversion of these instruments totals 4,033,838 shares (see Note 3, Note 4 and Note 7).
Fair Value of Financial Instruments
ASC Topic 820 defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
Included in the ASC Topic 820 framework is a three level valuation inputs hierarchy with Level 1 being inputs and transactions that can be effectively fully observed by market participants spanning to Level 3 where estimates are unobservable by market participants outside of the Company and must be estimated using assumptions developed by the Company. The Company discloses the lowest level input significant to each category of asset or liability valued within the scope of ASC Topic 820 and the valuation method as exchange, income or use. The Company uses inputs which are as observable as possible and the methods most applicable to the specific situation of each company or valued item.
Interest rate risk is the risk that the value of a financial instrument might be adversely affected by a change in the interest rates. The Company’s outstanding convertible notes payable issued in 2024 had fixed interest rates. Therefore, the Company was exposed to interest rate risk in that they could not benefit from a decrease in market interest rates. In seeking to minimize the risks from interest rate fluctuations, the Company manages exposure through its normal operating and financing activities.
Stock-based Compensation
The Company has a stock-based compensation plan, which is described in detail in Note 7 and Note 8, and records all stock-based payments, including grants of employee share options, at their fair values. The Company accounts for stock-based compensation instruments in accordance with the guidance promulgated under ASC 718, Compensation – Stock Compensation. The fair value of share options granted to employees and non-employees is estimated at the date of grant using the Black-Scholes option pricing model. The Company recognizes stock-based compensation expense over the requisite service period of the individual grants, which equals the vesting period, using the straight-line method or, in the case of performance based awards, based upon the terms of the performance conditions. Forfeitures, if any, are recorded as they occur. Any consideration paid by employees on exercising share options and the corresponding portion previously credited to contributed surplus are credited to share capital. The Black-Scholes option pricing model used by the Company to calculate option values was developed to estimate fair value.
F-12
Recent Accounting Pronouncements Not Yet Adopted
In November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses (“ASU 2024-03”). The standard requires more detailed disclosures about specified categories of expenses (including employee compensation, depreciation, and amortization) included in certain expense captions presented on the face of the income statement. ASU 2024-03 is effective for fiscal years beginning after December 15, 2026, and for interim periods within fiscal years beginning after December 15, 2027. Early adoption is permitted. The amendments may be applied either (1) prospectively to financial statements issued for reporting periods after the effective date of ASU 2024-03 or (2) retrospectively to all prior periods presented in the financial statements. The Company is currently evaluating the potential impact that adoption of this new standard will have on its consolidated financial statements and related disclosures.
The Company continually assesses any new accounting pronouncements to determine their applicability to the Company. Where it is determined that a new accounting pronouncement affects the Company’s financial reporting, the Company undertakes a study to determine the consequence of the change to its financial statements and assures that there are proper controls in place to ascertain that the Company’s financials properly reflect the change.
3. REVERSE RECAPITALIZATION
The Merger
On February 11, 2025, the Company, through its wholly owned subsidiary Adaptin Acquisition Co., consummated the Merger with Private Adaptin. Pursuant to the Merger, Private Adaptin was the surviving corporation and became a wholly owned subsidiary of Unite, and all of the outstanding common stock of Private Adaptin was converted into
The Merger was accounted for as a reverse recapitalization under U.S. GAAP. This determination was primarily due to the Company being determined to be a shell company in that it did not meet the U.S. GAAP definition of a business, did not have more than nominal assets, and did not have more than nominal operations at the time of the Merger. Under this method of accounting, the Company was treated as the “acquired” company for financial reporting purposes. Accordingly, the consolidated financial statements of the Company represent a continuation of the financial statements of Private Adaptin, with the Merger being treated as the equivalent of Private Adaptin issuing stock for the net assets of the Company, accompanied by a recapitalization. The net assets of the Company are stated at historical cost, with no goodwill or other intangible assets recorded and are consolidated with Private Adaptin’s financial statements on the Merger closing date. Results of operations prior to the Merger are presented as those of Private Adaptin. The shares and net loss per share, prior to the Merger, have been retroactively restated to reflect the common stock exchange ratio of
The Offering
On February 11, 2025, concurrent with the Merger, the Company issued, in a private placement offering (the “Offering”),
F-13
The offering period commenced on January 8, 2025 and was scheduled to continue until the later of (i) February 28, 2025, unless extended by the Company and the placement agent; (ii) the date on which the maximum offering amount of approximately $
In connection with the Offering, the placement agent and/or its subagents (a) were paid at each closing from the Offering proceeds a total cash commission of
In connection with the Merger, all officers and directors of the Company and their affiliates and associated entities entered into lock-up agreements with the Company for a term ending two years after the closing of the Merger, whereby they have agreed to certain restrictions on the sale or disposition (including pledge) of the Company common stock held by (or issuable to) them.
During the year ended December 31, 2025, in connection with the Offering, the Company issued
Conversion of Exchange Notes and 2024 Notes
At the Initial Closing, $
F-14
4. COMMON STOCK
See Note 3 - Reverse Recapitalization for details associated with the issuance of common stock and warrants, as well as details associated with the reverse recapitalization in connection with the Merger.
Common Stock Issuance
On April 2, 2025, in connection with the execution of a vendor contract, the Company issued
Follow-on Offering
On December 22, 2025, the Company issued, in a private placement offering (the “Follow-on Offering”) in an initial closing (the “Initial Closing”),
In connection with the Follow-on Offering, the Company paid: (a) to the placement agent a cash commission of
The A Warrants and B Warrants (see Note 3) contain certain anti-dilution provisions whereby the number of warrants and exercise price of warrants are subject to change if the Company sells its common stock at a price lower than the exercise price. The Follow-on Offering triggered the anti-dilution provision for the B Warrants and accordingly, the number of B Warrants increased to a total of
5. PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets are composed of the following as of December 31, 2025 and 2024:
| December 31, 2025 | December 31, 2024 | |||||||
| Prepaid insurance | $ | $ | ||||||
| Deferred research and development expenses | ||||||||
| Professional services retainers | ||||||||
| Other prepaid expenses | ||||||||
| $ | $ | |||||||
F-15
6. ACCRUED EXPENSES
Accrued expenses are composed of the following as of December 31, 2025 and 2024:
| December 31, | December 31, | |||||||
| 2025 | 2024 | |||||||
| Accrued research and development | $ | $ | ||||||
| Accrued professional fees | ||||||||
| Accrued consulting fees | ||||||||
| Accrued legal costs | ||||||||
| Accrued compensation | ||||||||
| Accrued directors fees | ||||||||
| Accrued royalties | ||||||||
| Accrued franchise taxes | ||||||||
| $ | $ | |||||||
7. STOCKHOLDERS’ DEFICIT
See Note 3 - Reverse Recapitalization for details associated with the issuance of common stock and warrants, as well as details associated with the reverse recapitalization in connection with the Merger.
See Note 4 – Common Stock for details associated with the issuance of common stock and warrants in the Company’s Follow-on Offering.
Warrants
See Note 3 - Reverse Recapitalization for details associated with the issuance of warrants, including their term and accounting treatment.
See Note 4 – Common Stock for details associated with the issuance of warrants, including their term and accounting treatment.
The Company estimated the fair value of the warrants granted using the Black-Scholes valuation model with the following assumptions:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Risk-free interest rate | ||||||||
| Expected term (years) | ||||||||
| Expected volatility | ||||||||
| Expected dividends | % | |||||||
F-16
The following table presents information related to warrants as of December 31, 2025:
| Class of Warrants | Quantity | Exercise Price | Expiration Date | |||||||
| Investor A | $ | |||||||||
| Investor B | $ | |||||||||
| Placement Agent | $ | |||||||||
| Investor A | $ | |||||||||
| Investor B | $ | |||||||||
| Placement Agent | $ | |||||||||
| Original | $ | |||||||||
| Exchange | $ | |||||||||
| Placement Agent | $ | |||||||||
| Total | ||||||||||
8. STOCK-BASED COMPENSATION
2025 Equity Incentive Plan
On February 11, 2025, the Company adopted the 2025 Equity Incentive Plan (the “2025 Plan”), which provides for the issuance of incentive awards of stock options, restricted stock awards, restricted stock units, stock appreciation rights and performance awards. The 2025 Plan was approved by the Company’s stockholders and Board of Directors on February 11, 2025 Awards under the 2025 Plan may be issued, at the discretion of the Board of Directors, to officers, key employees, consultants and directors of the Company and its subsidiaries.
The number of shares reserved for issuance under the 2025 Plan will increase, subject to approval by the board of directors, on February 11 of each of the years ending 2026 through 2035 by the number of shares equal to the lesser of
As of December 31, 2025, up to
Stock Options
On September 8, 2025, the Company granted stock options to purchase an aggregate of
The Company granted its Chief Financial Officer an option to purchase an aggregate of
The Company granted various directors and officers options to purchase an aggregate of
F-17
The Company granted a consultant (the “Consultant”) options to purchase an aggregate of
Of the Consultant Grant,
For the purposes of disclosure, as indicated above,
A summary of stock option activity during the year ended December 31, 2025, is as follows:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | Aggregate | ||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Options | Price | In Years | Value | |||||||||||||
| Outstanding, January 1, 2025 | $ | |||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Expired | ||||||||||||||||
| Forfeited | ||||||||||||||||
| Outstanding as of December 31, 2025 | $ | $ | ||||||||||||||
| Exercisable as of December 31, 2025 | $ | $ | ||||||||||||||
The Company estimated the fair value of the stock options granted using the Black-Scholes valuation model with the following assumptions:
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Risk-free interest rate | ||||||||
| Expected term (years) | ||||||||
| Expected volatility | ||||||||
| Expected dividends | ||||||||
F-18
The Company recognized stock-based compensation expense related to stock options of $
As of December 31, 2025, there was $
9. RELATED PARTY TRANSACTIONS
In connection with the Merger and Offering, the Company terminated the then-existing services agreement with Lucius Partners and fully repaid the unsecured promissory note of $
Following the Merger, the Company and Lucius Partners entered into a new professional services agreement whereby Lucius Partners agreed to provide advisory services to the Company for years following the Initial Closing (the “Advisory Period”). In connection with the agreement, the Company paid a cash fee of $
See Note 11 - Commitments and Contingencies - Executive Compensation for details associated with the forgiveness of debt by related parties.
10. FAIR VALUE
ASC 820, Fair Value Measurements and Disclosures (“ASC 820”), defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. ASC 820 requires disclosures about the fair value of all financial instruments, whether or not recognized, for financial statement purposes. Disclosures about the fair value of financial instruments are based on pertinent information available to the Company on December 31, 2025. Accordingly, the estimates presented in these consolidated financial statements are not necessarily indicative of the amounts that could be realized on disposition of the financial instruments. ASC 820 specifies a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect market assumptions. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement).
The three levels of the fair value hierarchy are as follows:
Level 1:
Inputs are unadjusted quoted prices in active markets for identical assets or liabilities available at the measurement date.
F-19
Level 2:
Inputs are observable unadjusted quoted prices for similar assets and liabilities in active markets, unadjusted quoted prices for identical or similar assets and liabilities in markets that are not active, inputs other than quoted prices that are observable, and inputs derived from or corroborated by observable market data.
Level 3:
Inputs are unobservable inputs which reflect the reporting entity’s own assumptions on what assumptions the market participants would use in pricing the asset or liability based on the best available information.
The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents, prepaid expenses, accounts payable, accrued expenses, notes payable and accrued interest approximate their fair market value based on the short-term maturity of these instruments.
Level 3 liabilities measured at fair value on a recurring basis include bifurcated embedded redemption features in convertible debt (see Note 3 - Reverse Recapitalization), which had a fair value of $
Liabilities measured at fair value on a recurring basis include bifurcated embedded redemption features in convertible debt (see Note 3) that no longer exist as of December 31, 2025. The following table is as of December 31, 2024:
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Derivative liabilities | $ | $ | $ | $ | ||||||||||||
The following table sets forth a summary of the changes in the fair value of Level 3 liabilities that are measured at fair value on a recurring basis during the years ended December 31, 2024 and 2025:
| 2024 | ||||
| Balance at January 1, 2024 | $ | |||
| Initial valuation of derivative liabilities for debt conversion | ||||
| Derivative liability established at debt extinguishment | ||||
| Change in fair value | ||||
| Balance at December 31, 2024 | $ | |||
| 2025 | ||||
| Balance at January 1, 2025 | $ | |||
| Change in fair value | ||||
| Extinguishment of convertible notes payable | ( | ) | ||
| Balance at December 31, 2025 | $ | |||
11. commitments and contingencies
Litigation
While there is currently no ongoing litigation, the Company may, from time to time, be involved in various legal matters that arise in the ordinary course of business. Should matters arise, management will then make a determination as to the ultimate disposition of these matters and measure if it could have a material adverse effect on the Company’s financial position, results of operations or liquidity.
F-20
License Agreement
In January 2023, the Company entered into a patent license agreement with Duke University and the National Cancer Institute, under the agency of the U.S. Department of Health and Human Services (the “Duke License”) for an exclusive, worldwide, sub-licensable license to the technology more fully described in Note 1. The Duke License was amended in August 2024 to include improvements within the definitions of patent rights and technical information. As a component of the Duke License, the Company agreed to make payments based on clinical and commercial milestones and continuing royalty payments on any sales made after approval by regulatory authorities. These milestones include initiation of Phase II or Phase III clinical trials, submission of applications for market approval in multiple jurisdictions including the U.S., EU and Japan and the initiation of post-approval commercial sales in the same jurisdictions. Based on an assumption that all milestones related to the current development program are met during the course of the Duke License, these milestone payments would total approximately $
The Company also agreed to pay royalties equal to low- to mid- single digit percentages of annual net sales on a country-by-country and product-by-product basis subject to downward adjustment to low single digit percentages of the Company’s net annual sales in the event there is no valid claim of a patent for the product, with minimum annual royalty levels established beginning in 2025. The Company also must pay Duke low to mid-double digit percentages of any sublicensing fees as set forth in the Duke License. Based on the minimum annual royalty levels established in the Duke License, the Company accrued $
Executive Compensation
On February 11, 2025, effective upon the closing of the Merger, the Company entered into executive employment agreements with three executive officers (each an “Executive” and collectively, the “Executives”). On October 3, 2025, the Company amended the employment agreements with one of the Executives to, among other things, amend the amount of time that Executive was going to devote to the Company and update their respective salary pro rata. The agreements include customary non-competition, non-solicitation, and confidentiality covenants; establish the Executives’ duties and compensation; and provide for their continued employment with the Company. The initial term of each of the employment agreements commenced upon the closing of the Merger and continues for terms ranging from to years, unless terminated sooner in accordance with the employment agreement. After the initial term expires, the employment agreements will automatically renew for successive one-year terms unless either the Company or the Executive provides written notice of their intent not to renew at least 90 days prior to the expiration of the then-current term.
The Company has agreed to pay the Executives annual base salaries of $
In the event that the Company issues additional securities, raising gross aggregate funds of $
The employment agreements may be terminated (a) automatically upon the Executive’s death; (b) by the Company upon disability, for cause or not for cause; or (c) by the Executive for good reason or no reason; all as defined in the employment agreements.
F-21
If the Company terminates the Executive’s employment without cause or if the Executive resigns for good reason, then the Executive will be entitled to separation benefits, consisting of
If the Company terminates the Executive’s employment without cause or if the Executive resigns for good reason, in connection with a change in control of the Company, then the Executive will be entitled to accelerated vesting of all equity awards, in addition to the separation benefits enumerated above.
On February 5, 2025, each of the Company’s Executives agreed to forever waive and discharge any obligation on the part of the Company to pay the consulting fees incurred and unpaid prior to consummation of the Merger. In the aggregate, the amount of consulting fees that were unpaid and waived under this agreement totaled $
12. INCOME TAXES
The Company reported a loss before income taxes of approximately $
The Company believes that there are no uncertain tax positions for which a liability (unrecognized tax benefit) should be recognized.
The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. During the years ended December 31, 2025 and 2024, the Company recognized no interest and penalties associated with unrecognized tax benefits. There are no tax positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within twelve months of the reporting date.
Under the tax statute of limitations applicable to the Internal Revenue Code, the Company and its U.S. subsidiary, either standalone or as part of the consolidated group, are subject to U.S. federal income tax examinations by the Internal Revenue Service for tax years 2022-2024. As the Company is carrying forward income tax attributes, such as net operating losses from 2022, these attributes can still be audited when utilized on returns filed in the future.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Due to the Company’s history of operating losses since inception, and the uncertainty of future taxable income, the Company has recorded a full valuation allowance against its deferred tax assets. As of December 31, 2024, the Company had recorded a valuation allowance against its deferred tax assets of approximately $
As of December 31, 2025 and 2024, the Company had U.S. federal NOL carryforwards of $
The NOL carryforwards are subject to review and adjustment by the U.S. and state tax authorities. NOL carryforwards may become subject to an annual limitation in the event of certain cumulative changes in ownership interest of significant shareholders, as defined under Section 382 of the Internal Revenue Code. This could limit the amount of NOLs that the Company can utilize annually to offset future taxable income or tax liabilities. As of December 31, 2025, the Company has not performed such an analysis evaluating the potential limitations of the Company’s NOL carryforwards due to the “change in ownership” provisions as defined under Section 382. Subsequent ownership changes and proposed future changes to tax laws in respect of the utilization of losses carried forward may further affect the limitation in future years.
The Company may also be eligible for federal and state research and development (“R&D”) tax credits; however, as of December 31, 2025, no formal R&D credit study has been completed, and no related credits have been recorded due to cumulative NOLs and the full valuation allowance position. Any R&D credits identified may be subject to annual limitations under Section 383 of the Internal Revenue Code in the event of a change in ownership.
F-22
On July 4, 2025, the One Big Beautiful Bill Act (“OBBBA”) was signed into law, introducing significant and wide-ranging changes to the U.S. federal tax system. The OBBBA did not have a material impact on income tax benefit or expense or related tax assets or liabilities given that the Company remains in a net operating loss (“NOL”) position and has previously recorded a fully-offsetting valuation allowance against all deferred tax assets, primarily related to the prior capitalization and amortization of domestic research and development expenses. The Company consulted with its tax advisors and reviewed the various elections available under the OBBBA for the treatment of domestic research and development expenses and, as a result, fully expensed domestic research and development costs in 2025 as well as accelerating the amortization of previously capitalized and unamortized research and development costs. The Company did not apply retroactive treatment to any prior tax periods.
The Company’s net deferred tax assets are as follows:
| As of December 31, | ||||||||
| Deferred tax assets: | 2025 | 2024 | ||||||
| Net operating loss carryforward - Federal | $ | $ | ||||||
| Timing difference - §174 R&D costs | ||||||||
| Timing difference - Stock-based compensation | ||||||||
| Timing difference - Accrued Consulting | ||||||||
| Total deferred tax assets | ||||||||
| Less - Valuation allowance | ( | ) | ( | ) | ||||
| Net deferred tax assets | $ | $ | ||||||
Upon adoption of ASU No. 2023-09, for the year ended December 31, 2025, the provision for income taxes differs from the expense that would be obtained by applying the U.S. federal statutory income tax rate as a result of the following:
| 2025 | ||||||||
| Amount | Percent | |||||||
| U.S. statutory rate[1] | ( | ) | % | |||||
| Changes in valuation allowance | - | % | ||||||
| Deferred changes | ( | ) | % | |||||
| Non-taxable or non-deductible expenses | - | % | ||||||
| Other | - | % | ||||||
| Effective tax rate | % | |||||||
| (1) |
Prior to the adoption of ASU No. 2023-09, for the year ended December 31, 2024, the provision for income taxes differs from the expense that would be obtained by apply the U.S. statutory income tax rate as result of the following:
| 2024 | ||||||||
| Amount | Percent | |||||||
| U. S. Statutory Rate | ( | ) | % | |||||
| Statutory state rate, net of federal benefit | ( | ) | % | |||||
| Non-taxable or non-deductible expenses | ( | ) | % | |||||
| Change in valuation allowance | - | % | ||||||
| Income tax expense (recovery) reported in the consolidated statement of loss | % | |||||||
F-23
13. SUBSEQUENT EVENTS
Follow-on Offering
Subsequent to December 31, 2025, the Company completed a secondary closing under its Follow-on Offering (the “February 2026 Closing”), issuing
The February 2026 Closing triggered the anti-dilution provision for the B Warrants (see Note 3) and accordingly, the number of B Warrants increased to a total of
Subsequent to December 31, 2025, the Company completed a third closing under its Follow-on Offering (the “March 2026 Closing”), issuing
The March 2026 Closing triggered the anti-dilution provision for the B Warrants (see Note 3) and accordingly, the number of B Warrants increased to a total of
A Warrant Expiration Date
Subsequent to December 31, 2025, the Company had not yet been approved for its ticker symbol allowing it to register and trade on the OTC market. As such, in accordance with the terms of the A Warrant (see Note 3), the expiration date of these warrants was extended by six months from March 31, 2026 until September 30, 2026.
Option Grants and Modifications
Subsequent to December 31, 2025, the Company’s Board of Directors approved a modification to certain stock options granted to a consultant in 2025, converting performance-based vesting conditions to service-based vesting over 48 months. In addition, the Company’s Board of Directors approved an option grant to a new consultant that was engaged in March 2026 for
F-24
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
There are not and have not been any disagreements between the Company and WithumSmith+Brown, PC on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, or any reportable event as described in paragraph (a)(1)(v) of Item 304 of Regulation S-K.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
Disclosure controls are procedures that are designed with the objective of ensuring that information required to be disclosed in our reports filed under the Exchange Act, such as this Report, is recorded, processed, summarized and reported within the time period specified in the SEC’s rules and forms. Disclosure controls are also designed with the objective of ensuring that such information is accumulated and communicated to our management, including the President and Chief Executive Officer (our principal executive officer) and Chief Financial Officer (our principal financial and accounting officer), as appropriate to allow timely decisions regarding required disclosure.
In connection with the preparation of this Report, management, with the participation of our principal executive officer and principal financial and accounting officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures as of December 31, 2025, pursuant to Rule 13a-15(b) under the Exchange Act. Based upon that evaluation, our principal executive officer and principal financial and accounting officer concluded that, as of December 31, 2025, our disclosure controls and procedures were not effective to ensure appropriate review of the internally prepared financial statements including the lack of adequate segregation of accounting functions which could have resulted in material misstatements in the financial statements
We performed additional analysis as deemed necessary to ensure that our financial statements were prepared in accordance with U.S. generally accepted accounting principles. Accordingly, management believes that the financial statements included in this Report present fairly in all material respects our financial position, results of operations and cash flows for the period presented.
Management’s Report on Internal Controls Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such terms are defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act. Our internal control system was designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes, in accordance with generally accepted accounting principles. Because of inherent limitations, a system of internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate due to change in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2025. Management based this assessment on criteria for effective internal control over financial reporting described in “Internal Control - Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of its internal control over financial reporting. Management reviewed the results of its assessment with our Board of Directors.
26
Based on this assessment, management concluded that our internal control over financial reporting was not effective as the previously disclosed material weaknesses in our internal control over financial reporting continued to exist as of December 31, 2025. The following material weaknesses in our internal control over financial reporting were present as of December 31, 2024 and continued to exist as of December 31, 2025: errors in accounting for non-routine transactions; errors in accounting for prepaid and accrued research and development costs; and errors in the accounting for tax provisions. The material weaknesses identified were a result of insufficient internal resources to design, implement, document, and operate effective internal controls around our financial reporting process.
This Report on Form 10-K does not include an attestation report of our independent registered public accounting firm, regarding internal controls over financial reporting. Our internal control over financial reporting was not subject to such attestation as we are a “smaller reporting company” as defined by Item 10 of Regulation S-K.
Changes in Internal Control over Financial Reporting
In an effort to remediate the material weaknesses described above, the Company has added and is seeking additional qualified accounting and financial reporting personnel, in addition to designing and implementing financial reporting systems, processes, policies and internal control. Such efforts are incomplete as of December 31, 2025.
These changes in our internal control over financial reporting were identified in connection with the evaluation required by paragraph (d) of Rule 13a-15 or 15d-15(f) under the Exchange Act that occurred during the period covered by this Report and have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations of the Effectiveness of Control
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations of any control system, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected.
ITEM 9B. OTHER INFORMATION.
No director or officer of the Company
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
Not applicable.
27
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The following table provides information regarding Adaptin’s executive officers and directors as of December 31, 2025:
| Name | Age | Positions | ||
| Executive Officers | ||||
| Michael J. Roberts, PhD | 56 | Chief Executive Officer and Director | ||
| Simon C. Pedder, PhD | 65 | Executive Chairman and Director | ||
| Timothy L. Maness, CPA | 65 | Chief Financial Officer | ||
| L. Arthur Hewitt, PhD | 72 | Chief Development Officer | ||
| Non-Employee Directors | ||||
| Patrick Gallagher(1) | 61 | Director | ||
| Anthony Zook(1) | 65 | Director | ||
| J. Nick Riehle(1) | 73 | Director |
| (1) | Member of the Audit Committee. |
Executive Officers
Michael J. Roberts, PhD - Chief Executive Officer and Director
Dr. Roberts has been our Chief Executive Officer and a member of our board of directors since March 2021. He has over 25 years of pharmaceutical research, development, corporate development, and executive experience. He is co-founder and President and CEO of Adaptin Bio and also serves on the board of directors. Prior to Adaptin he was co-founder of Corino Therapeutics and is currently its acting CEO. Dr. Roberts led and ran all activities related to Corino and the development of a disease-modifying drug called CRX-1008, which is used to treat the protein disorder transthyretin amyloidosis. He is a pharmaceutical and biotech consultant and owner of MAC B Consulting. Prior to Corino, Dr. Roberts was VP, Business Development and Corporate Officer of publicly traded Chelsea Therapeutics International, Ltd. He was responsible for business development efforts focused primarily on licensing and M&A with consideration to pipeline management. He led the sale of Chelsea Therapeutics to H. Lundbeck A/S in 2014 for approximately $658 million. Prior to Chelsea Therapeutics, Dr. Roberts was Director of Business Development for their Nektar Therapeutics’ Molecule Engineering technology and completed a number of transactions with large and specialty pharmaceutical companies. Dr. Roberts has completed pharmaceutical transactions valued at over $1B. Prior to this he was Manager of Biopharmaceutical Research at Shearwater Corporation where he led and was successful in the development of preclinical drug candidates from initial stages of research through Phase I clinical study, including inventing the product Movantik™, a treatment for opioid-induced constipation, subsequently licensed to AstraZeneca in a transaction valued at approximately $1 billion. Shearwater was sold to Inhale Therapeutic Systems (now known as Nektar Therapeutics) in 2001 for approximately $200 million. Dr. Roberts obtained his Ph.D. in Materials Science from the University of Alabama and B.S. in Chemical Engineering from Pennsylvania State University. We believe that Dr. Roberts’ experience in the life sciences industry and in the operation of publicly traded companies as an executive qualifies him to serve on our board of directors.
Simon C. Pedder, PhD - Executive Chairman and Director
Dr. Pedder has been our Executive Chairman and a member of our board of directors since October 2023. He has a career of over 30 years in drug development and commercialization. He was Chief Executive Officer of Nirogy from November 2021 to November 2022. From December 2016 to November 2021, he had leadership roles as Chief Business and Strategy Officer for Athenex; President and CEO of Cellectar Biosciences from April 2014 to June 2015; President and CEO of Chelsea Therapeutics International, Ltd. from May 2004 to July 2012; and Global Vice President of Oncology Pharma Business and Executive Officer at Hoffmann-LaRoche. Previous positions at Roche included Life Cycle Leader and Global Project Leader of Pegasys/IFN and Head of Hepatitis Franchise. Prior to that, he was Clinical Leader for a number of development compounds at Roche. Dr. Pedder served on the board of directors of Cerecor, Inc. from April 2018 to June 2020, Mateon Therapeutics, Inc. from March 2016 to April 2019 and Delcath Systems, Inc. from November 2017 to April 2019.
28
Early in his career, he was a faculty member in the Department of Pharmacology in College of Medicine at the University of Saskatchewan, where he obtained his Ph.D. in Clinical Pharmacology. During his longstanding career in pharmaceutical development, Dr. Pedder played key roles in the successful development and commercialization of multiple proprietary pharmaceutical products including Tasmar®, Pegasys®, Copegus®, Northera® and Klisyri®. In addition to his Ph.D., Dr. Pedder obtained a Master of Science in Toxicology from Concordia University, a Joint Honors Bachelor degree in Environmental Studies/Biology from the University of Waterloo, and he completed the Roche-sponsored Pharmaceutical Executive Management Program at Columbia Business School. We believe Dr. Pedder’s experience in the life sciences industry and in the operation of publicly traded companies as an executive and board member qualifies him to serve on our board of directors.
Timothy L. Maness, CPA, CGMA - Chief Financial Officer
Mr. Maness has been our Chief Financial Officer since September 2024. Since April 2016, he had provided financial consulting services through Adamanteus, LLC. Mr. Maness has been a consultant to multiple publicly held and private-equity-backed life sciences companies, and he has held several senior financial management roles.
In 2018, Mr. Maness was engaged by Milestone Pharmaceuticals to prepare and lead its initial public offering, serving as its interim CFO, CAO, and Vice President of Finance. Before that, he served as the CFO of Ballantyne Therapeutics. Earlier, during his eight years as the Senior Director of Finance and Corporate Controller at Chelsea Therapeutics International, he was heavily involved in Chelsea’s reverse-public-shell merger to complete its public listing in 2005. Mr. Maness is a noted expert in forensic accounting and internal audit and has served as the internal auditor hired by public company audit committees on several occasions.
Mr. Maness received a Bachelor of Science in Accounting from the University of North Carolina at Charlotte, and he is licensed as a Certified Public Accountant and as a Chartered Global Management Accountant.
L. Arthur Hewitt, PhD - Chief Development Officer
Dr. Hewitt has been our Chief Development Officer since September 2024. He has worked for approximately 35 years in clinical research and regulatory affairs. From December 2016 to December 2021, he served as a scientific advisor to Amneal Pharmaceuticals and Senior Scientific Advisor - Neurology at Lundbeck Pharmaceuticals. Prior to Lundbeck, Dr. Hewitt was Chief Scientific Officer for Chelsea Therapeutics International, Ltd. from January 2010 to June 2014 and Vice President, Drug Development from May 2004 to June 2014. At Chelsea, he oversaw the clinical development and regulatory approval of Northera™ (droxidopa), a novel therapeutic agent, for the treatment of symptomatic neurogenic orthostatic hypotension, or Neurogenic OH, in patients with primary autonomic failure (Parkinson’s disease, or PD, multiple systems atrophy, or MSA, and pure autonomic failure, or PAF), dopamine β-hydroxylase, or DBH, deficiency and non-diabetic autonomic neuropathy. Prior to Chelsea, Dr. Hewitt served as an independent contractor from January 2003 to May 2004, and as Director of Scientific Affairs at Shearwater Corporation, a drug delivery company, from October 2002 until January 2003.
From July 1991 until November 2000, Dr. Hewitt was Director of Scientific Affairs for Amgen Canada where he oversaw the clinical research and regulatory requirements for a wide variety of proprietary biologic treatments undergoing Phase I, II and III research. Thirteen individual investigational new drug, or IND, research programs were established covering the therapeutic domains of Hematology, Oncology, Neurology, Infectious Disease, and Inflammation. Dr. Hewitt also developed clinical research programs supporting the approval of three products including Neupogen, Stemgen and Infergen and six supplementary approvals. Prior to Amgen, Dr. Hewitt held positions at Jansen Pharmaceuticals and Park-Davis where he developed research programs for multiple neurology and oncology products. Prior to entering the pharmaceutical industry, he was a Lecturer at Concordia University in Montreal, Canada. Dr. Hewitt obtained his Ph.D. in Pharmacology from the University of Montreal. Additionally, Dr. Hewitt received a Master of Science degree in Toxicology and a B.Sc. (Hon) in Comparative Anatomy and Physiology from Concordia University.
29
Non-Employee Directors
Patrick Gallagher
Mr. Gallagher has been a member of our board of directors since February 2025. He has 20 years of healthcare experience including alternative investments, research and marketing in both the public and private markets. Since January 2018, he has served as the Chief Executive Officer and as a member of the board of directors of Voltron Therapeutics, a privately held biotechnology company. Since August 2014, Mr. Gallagher has served as a Managing Director of Laidlaw & Company (UK) Ltd. From January 2018 to September 2024, Mr. Gallagher served as the Chief Executive Officer and a member of the board of directors at PD Theranostics, Inc. He has also served as Treasurer of Aerwave Medical, Inc. since November 2020. Formerly, Mr. Gallagher was a founding partner and Chief Executive Officer of BDR Research Group, LLC (“BDR”), from July 2001 through October 2010. BDR was an independent sell-side research firm specializing in healthcare investing, financing and operations, serving the institutional investing community at large.
Mr. Gallagher served as VP of Business Development and Investor Relations as well as a strategic consultant for Kinex Pharmaceuticals, a biotechnology firm focused on next-generation therapies in oncology and immunology (now traded on NASDAQ: ATNX). He also served as an advisor to CHD Biosciences, a novel antimicrobial company from July 2012 through August 2014. Mr. Gallagher has served on the boards of directors of BioSig Technologies, Inc., NASDAQ-listed a medical device company that is developing a proprietary technology platform in the electrophysiology space since July 2014 Cingulate Therapeutics, a therapeutics company with a novel drug delivery platform since January 2014; Evermore Global, a global special situations money manager since June 2015; and Algorithm Sciences, Inc. since May 2019. Since September 2014, Mr. Gallagher has served as a Managing Partner at Laidlaw Venture Partners dba Laidlaw & Company (UK) Ltd. and is part of the Investment Banking Healthcare Team. Mr. Gallagher also is a member of Lucius Partners.
Mr. Gallagher earned his Master in Business Administration from Penn State University and his Bachelor of Science in Finance from the University of Vermont. We believe that Mr. Gallagher’s extensive experience in the life sciences industry qualifies him to serve on our board of directors.
Anthony Zook
Mr. Zook has been a member of our board of directors since February 2025. He currently is a member of Lucius Partners. Since July 2020, Mr. Zook has served as a director of BioSig Technologies, Inc. (Nasdaq: BSGM). From May 2023 to December 2024, Mr. Zook was a director of NeoGenomics (Nasdaq: NEO), an oncology laboratory service company where he also served as the chair of the compensation committee. From January 2022 to December 2024, he also served as Chairman to Voltron Therapeutics and Algorithm Sciences, respectively. Mr. Zook was executive vice president of global commercial operations of AstraZeneca Plc from 2010 until 2012. He also served as president and chief executive officer of the North American division of AstraZeneca Plc from 2006 until 2009, and president of Medimmune, the company’s wholly owned biologics division from 2009 until 2010. Mr. Zook previously served as a member of the board of directors of AltheRx from 2013-2014, InHibikase in 2014, Rib-X Pharmaceuticals in 2009, the National Pharmaceutical Council from 2007-2009, PhRMA from 2011-2012, the Pennsylvania Division of the American Cancer Society from 2005-2007 and his alma mater, Frostburg State University from 2016-2018 and re-joined in 2021, where he earned a B.S. degree. Mr. Zook also earned an A.A. degree in chemical engineering from Pennsylvania State University. We believe Mr. Zook’s extensive commercialization experience and expertise in executive leadership qualify him to serve on our board of directors.
J. Nick Riehle
Mr. Riehle has been a member of our board of directors since February 2025. He has over 25 years of business and management experience with both large companies and start-up ventures, and 18 years of that has been as a chief financial officer of pharmaceutical and software companies, including taking these same companies public. Mr. Riehle formerly served as the CFO of Athenex, Inc., managing all aspects of the accounting, treasury, IT, HR, and legal functions and providing support to the company when it went public in June 2017. Prior to this, he was a freelance contractor for companies seeking his expertise in financing and related support. From 2004 - 2014, Mr. Riehle was the CFO of Chelsea Therapeutics International, Ltd. He managed all aspects of the accounting, treasury, facilities, IT, HR, IR and legal functions, including supporting the company in over $300 million in public and private equity financings and being actively involved in the sale of the company to H. Lundbeck A/S in 2014 for $658 million. Prior to Chelsea Therapeutics, Mr. Riehle was the CFO for HAHT Commerce, Inc., managing all aspects of treasury, accounting, IT, legal and inventory administration, as well as having significant involvement in sales administration, services management, HR, facilities and related operational activities, including supporting the company in transactions involving over $60 million in venture financing. Prior to HAHT Commerce, Mr. Riehle held various positions in Canada, the United States and Asia with Nortel Networks, including financial planning and analysis, accounting, marketing and sales administration, government relations, and general administration.
30
Mr. Riehle earned his Bachelor of Commerce from McGill University, his MBA from York University and the Certified Management Accountant (CMA) designation in Ontario, Canada. We believe that Mr. Riehle’s experience in the life sciences industry qualifies and in the operation of publicly traded companies as a senior financial executive qualifies him to serve on our board of directors.
Corporate Governance
Appointment of Officers
Our executive officers are appointed by, and serve at the discretion of, our board of directors provided, however, that the board of directors may empower the Chief Executive Officer of the Company to appoint any officer other than the Executive Chairperson, the Chief Executive Officer, the President, the Chief Financial Officer or the Treasurer.
Board of Directors Composition
Our board of directors currently consists of five members. Dr. Roberts, Dr. Pedder, Mr. Gallagher, Mr. Zook, and Mr. Riehle have been designated to serve as members of our board of directors.
Each of our current directors will continue to serve until the election and qualification of his successor, or his earlier death, resignation, disqualification or removal.
Director Independence
Our securities are not listed on a national securities exchange or on any inter-dealer quotation system that has a requirement that a majority of directors be independent. We evaluate independence by the standards for director independence set forth in the Nasdaq Marketplace Rules. Under such rules, our board of directors has determined that all members of the board of directors except for Dr. Roberts and Dr. Pedder are independent directors under Nasdaq Listing Rule 5605(a)(2). In making such independence determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances that our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director.
Under Nasdaq Marketplace Rules, a director only qualifies as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. In order to be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (i) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries; or (ii) be an affiliated person of the listed company or any of its subsidiaries. We intend to satisfy the audit committee independence requirements of Rule 10A-3 as of the time we list on a national securities exchange.
Our board of directors has undertaken a review of the independence of each director and considered whether each director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. As a result of this review, our board of directors determined that Messrs. Gallagher, Riehle and Zook are “independent directors” as defined under the applicable rules and regulations of the SEC and the listing requirements and rules of Nasdaq. In making these determinations, our board of directors reviewed and discussed information provided by the directors and us with regard to each director’s business and personal activities and current and prior relationships as they may relate to us and our management, including the beneficial ownership of our capital stock by each non-employee director and the transactions involving them described in the section titled “Certain Relationships and Related Party Transactions.”
31
Family Relationships
There are no family relationships by between or among the members of the board of directors or other executive officers of the Company.
Legal Proceedings
None of the Company’s directors or executive officers are involved in any legal proceedings described in Item 401(f) of SEC Regulation S-K.
Code of Ethics
The Company has adopted a Code of Ethics and Business Conduct that applies to all officers, directors and employees (the “Code of Ethics”). The Code of Ethics is available under the heading “Governance Documents” on the Company’s website at ir.adaptinbio.com. If the Company makes any substantive amendments to the Code of Ethics or grants any waiver from a provision of the Code of Ethics to any executive officer or director, the Company will promptly disclose the nature of the amendment or waiver on its website.
The Code of Ethics addresses, among other matters, conflicts of interest and corporate opportunities, fair dealing, record- keeping and public disclosures, compliance with laws and corporate policies, confidentiality and corporate assets, and reporting and consequences of violations. The provisions of the Code of Ethics are intended to reflect current best practices and enhance the Company’s personnel’s understanding of the Company’s standards of ethical business practices, promote awareness of ethical issues that may be encountered in carrying out an employee’s or director’s responsibilities and improve clarity as to how to address ethical issues that may arise.
Insider Trading
The Company has
Committee of the Board of Directors
Our board of directors has an Audit Committee, which has the composition and responsibilities described below. Members serve on the Audit Committee until their resignation or until otherwise determined by our board of directors.
Audit Committee
The members of our Audit Committee are Messrs. Gallagher, Riehle, and Zook. Mr. Riehle serves as the Chair of this committee. The board of directors has determined that Mr. Riehle is “independent” under the Nasdaq Marketplace Rules and standards established by the SEC rules regarding audit committee members as set forth above. The board of directors has determined that Mr. Riehle qualifies as an “audit committee financial expert” as defined by applicable SEC rules.
32
The Audit Committee oversees on behalf of the board of directors (a) the conduct of the Company’s accounting and financial reporting processes, the audits of our financial statements and the integrity of the Company’s audited financial statements and other financial reports; (b) the performance of the Company’s internal accounting, internal auditing, and financial controls function; (c) the engagement, replacement, compensation, qualifications, independence and performance of our independent auditors, and (d) the portions of our Code of Conduct and Ethics and related policies regarding our accounting, internal accounting controls or auditing matters. The Audit Committee also reviews and approves or disapproves related party transactions identified in Item 404 of SEC Regulation S-K and makes recommendations to the full board of directors regarding the same.
The Audit Committee meets privately with our independent registered public accounting firm from time to time, and such firm has unrestricted access to, and reports directly to, the Audit Committee.
ITEM 11. EXECUTIVE COMPENSATION.
Information with respect to the Company’s directors and executive officers is described in the section titled “Management” above.
Director Compensation
Following the Merger, the Company provides annual cash compensation of $30,000 to its non-employee directors who are not otherwise paid by a major investor in or advisor to the Company (“Independent Directors”). The chair of the Audit Committee receives an additional $15,000 in annual cash compensation. Independent Directors will also be eligible to receive such equity grants or awards as may be approved by the board of directors in its discretion. The Company has granted each Independent Director with equity awards equal to 1.0% of the Company’s fully diluted shares outstanding at the time of grant. Drs. Roberts and Pedder are employees and executive officers of the Company and are not separately compensated for their service as directors.
Executive Officer Compensation
This section discusses the material components of the executive compensation program for the Company’s named executive officers (“NEOs”) who appear in the “Summary Compensation Table” below. In 2025, the “named executive officers” and their positions with the Company were as follows:
| ● | Michael J. Roberts: President and Chief Executive Officer |
| ● | Simon C. Pedder: Executive Chairman |
| ● | Timothy L. Maness: Chief Financial Officer |
This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs. Actual compensation programs that we adopt may differ materially from the currently planned programs summarized in this discussion.
33
Summary Compensation Table
The following table sets forth information concerning the compensation of the named executive officers for the Company’s two most recent fiscal years.
| Name and principal position | Year | Salary | Bonus | Stock awards | Option awards(4) | All other compensation | Total | |||||||||||||||||||||
| Simon C. Pedder, PhD | 2025 | 210,000 | -- | -- | 400,600 | $ | - | $ | 610,600 | |||||||||||||||||||
| Executive Chairman | 2024 | -- | -- | -- | -- | $ | 40,000 | (1) | $ | 40,000 | ||||||||||||||||||
| Michael J. Roberts, PhD | 2025 | 420,000 | -- | -- | 915,700 | $ | - | $ | 1,335,700 | |||||||||||||||||||
| Chief Executive Officer | 2024 | -- | -- | -- | -- | $ | 60,000 | (2) | $ | 60,000 | ||||||||||||||||||
| Timothy L. Maness, CPA | 2025 | 237,500 | -- | -- | 392,500 | $ | - | $ | 630,000 | |||||||||||||||||||
| Chief Financial Officer | 2024 | -- | -- | -- | -- | $ | 20,550 | (3) | $ | 20,550 | ||||||||||||||||||
| (1) | Represents amount paid to Dr. Pedder pursuant to a compensation agreement prior to his employment by the Company. |
| (2) | Represents amount paid to MAC B Consulting LLC, which is owned by Dr. Roberts, pursuant to a compensation agreement prior to his employment by the Company. |
| (3) | Represents amount paid to Adamanteus LLC, which is owned by Mr. Maness, pursuant to a consulting agreement prior to his employment by the Company. |
| (4) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the option awards granted during the applicable fiscal year, computed in accordance with ASC 718 for stock-based compensation transactions. Assumptions used in the calculation of these amounts are included in Note 8 to our audited consolidated financial statements included in our Annual Report on Form 10-K. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the share options, the exercise of the share options, or the sale of the common shares underlying such share options. |
Non-Equity Incentive Plan Compensation
In accordance with the terms of their employment agreements, as amended, our named executive officers are eligible to receive annual bonuses of up to a percentage of each executive’s gross base salary based on individual performance, company performance or as otherwise determined appropriate by our Board.
2025 Annual Bonus Information
| Target Bonus | Target Bonus | Actual Bonus | ||||||||||
| Name | % of Salary | $ | $ | |||||||||
| Simon C. Pedder, PhD | 30 | % | $ | 63,000 | $ | - | ||||||
| Michael J. Roberts, PhD | 50 | % | $ | 210,000 | $ | - | ||||||
| Timothy L. Maness, CPA | 30 | % | $ | 71,250 | $ | - | ||||||
For the year ended December 31, 2025, no bonuses will be paid to our NEOs. For fiscal year 2026, the Board of Directors decided to maintain the same target annual bonus opportunities for all NEOs.
Equity based Incentive Awards
The Company adopted an Equity Incentive Plan (the “2025 EIP) on February 11, 2025 in conjunction with the Merger. Each Executive will be eligible to receive such equity grants or awards as may be approved by the board of directors in its discretion.
34
Subject to approval by the board of directors and the terms of the Company’s equity compensation plan then in place, in the event the Company issues additional securities raising aggregate funds of $10,000,000 (in one or more transactions), occurring, if at all, within two years following the Merger (the “Additional Financing Period”), the Company will grant each Executive options to purchase a number of shares of Common Stock of the Company (the “Anti-Dilution Options”) sufficient to ensure that their respective ownership immediately following the Additional Financing Period, on a fully diluted basis and assuming the exercise of all outstanding options (whether or not then exercisable) is equal to their respective ownership immediately following the Merger, as determined on a fully diluted basis and assuming the exercise of all outstanding options (whether or not then exercisable). The per share exercise price of the Anti-Dilution Options will be equal to the fair market value of a share of the Company’s Common Stock on the date of grant, as determined by the board of directors. The Anti-Dilution Options, if any, will become exercisable in four equal annual installments, in each case subject to the continued employment of each Executive with the Company on the date each such vesting milestone is achieved, and will be subject to the terms of the Company’s equity incentive plan then in place and a related option grant agreement to be entered between Executive and the Company.
Outstanding Equity Awards at Fiscal Year-End
As of the end of our most recently completed fiscal year, our named executive officers held the following outstanding equity awards.
| Name and principal position | Grant Date | Vesting Commencement Date | Number of Securities Underlying Unexercised Options (#) (Exercisable) | Number of Securities Underlying Unexercised Options (#) (Unexercisable) | Option Exercise Price | Option Expiration Date | ||||||||||||
| Simon C. Pedder, PhD | 9/8/2025 | 9/8/2025 | 16,019 | 176,212 | $ | 4.40 | 9/7/2035 | |||||||||||
| Executive Chairman | ||||||||||||||||||
| Michael J. Roberts, PhD | 9/8/2025 | 9/8/2025 | 36,616 | 402,770 | $ | 4.40 | 9/7/2035 | |||||||||||
| Chief Executive Officer | ||||||||||||||||||
| Timothy L. Maness, CPA | 9/8/2025 | 9/8/2025 | 104,125 | 88,106 | $ | 4.40 | 9/7/2035 | |||||||||||
| Chief Financial Officer | ||||||||||||||||||
Policies and Practices Related to Stock Option Grants
The following discussion of the timing of option awards in relation to the disclosure of material nonpublic information is provided as required by Item 402(x) of Regulation S-K. The Company does not have a written policy regarding the timing of option awards in relation to the disclosure of material nonpublic information. In the event stock options are issued, the Compensation Committee does not intend to take material nonpublic information into account when determining the timing and terms of option awards. The Company has not timed the disclosure of material nonpublic information to affect the value of executive compensation.
Employment Agreements with Named Executive Officers
We entered into executive employment agreements with each of Dr. Roberts, Dr. Pedder, and Mr. Maness (each an “Executive” and collectively, the “Executives”), which were effective upon the closing of the Merger. Our agreement with Mr. Maness was subsequently amended on October 3, 2025. The agreements include customary non-competition, non-solicitation, and confidentiality covenants; establish the Executives’ duties and compensation; and provide for their continued employment with the Company. The discussion that follows summarizes certain other terms of these agreements.
35
Term. The initial term of each of the employment agreements commenced upon the closing of the Merger and continue for a term of three years in the case of Dr. Roberts and Dr. Pedder, and two years in the case of Mr. Maness, unless terminated sooner in accordance with the employment agreement (see “Termination” below). After the initial term expires, the employment agreements will automatically renew for successive one-year terms unless either the Company or the Executive provides written notice of their intent not to renew at least 90 days prior to the expiration of the then-current term.
Board Service. In the case of Drs. Roberts and Pedder, the Company will use commercially reasonable efforts to cause these individuals to be elected as members of the Company’s board of directors throughout the terms of their employment agreements.
Base Salary. The Company is currently paying each Executive a base salary at the following annual rates:
Dr. Roberts - $480,000
Dr. Pedder - $240,000
Mr. Maness - $300,000
These salaries will be reviewed by the board of directors from time to time and may be increased in the board of directors’ sole discretion. Salaries may not be reduced except in connection with an across-the-board reduction of executive-level salaries in which the Executive will not be subject to a greater reduction, on a percentage basis, than any other executive- level employee.
Other Benefits. Dr. Roberts and Dr. Pedder will each be entitled to no less than 25 paid vacation days per year. Mr. Maness will be entitled to no less than 20 paid vacation days per year. The Executives will also be entitled to such other benefits, and to participate in such benefit plans, as are generally made available to similarly situated senior executive employees of the Company. The Company will also reimburse the Executives for all reasonable business expenses they incur in connection with the performance of their duties.
Termination. The employment agreements may be terminated in the following circumstances:
| ● | Automatically effective upon the Executive’s death. |
| ● | By the Company upon notice to the Executive in the event of the Executive’s disability. “Disability” means the inability of the Executive to perform the essential functions of their job for 90 consecutive days or for 120 days in any one-year period due to the condition of the Executive’s physical, mental, or emotional health. |
| ● | By the Company for cause. “Cause” means the Executive’s (i) fraud, embezzlement or misappropriation with respect to the Company; (ii) willful or grossly negligent misconduct that has or may reasonably be expected to have a material adverse effect on the property, business, or reputation of the Company; (iii) material breach of their employment agreement; (iv) willful failure or refusal to perform their material duties or willful failure to follow any specific lawful instructions of the board of directors (in the case of Dr. Roberts and Dr. Pedder) or the chief executive officer (in the case of Mr. Maness); (v) conviction or plea of nolo contendere in respect of a felony or of a misdemeanor involving moral turpitude; or (vi) material failure to comply with the Company’s workplace rules, policies, or procedures. |
| ● | By the Company for any reason other than for cause or the Executive’s disability. |
| ● | By the Executive for good reason. “Good reason” means the occurrence of any of the following events without the Executive’s written consent: (i) the Company’s requiring the Executive to be based at any office or location more than 25 miles from their principal work location, except for travel reasonably required in the performance of the Executive’s responsibilities to the Company; (ii) a material reduction of the Executive’s base salary not in compliance with their employment agreement; (iii) a material diminution of the Executive’s authority, duties, or responsibilities; or (iv) the Company’s material breach of the Executive’s employment agreement. The Company will have an opportunity to cure any such conditions following notice by the Executive. |
| ● | By the Executive upon 30 days’ written notice to the Company at any time for any reason. |
36
Separation Benefits Not in Connection with a Change in Control. If the Company terminates the Executive’s employment without cause or if the Executive resigns for good reason, in either case not in connection with a change in control of the Company, then the Executive will be entitled to the following benefits:
| ● | 24 months of then-current base salary in the case of Dr. Roberts and Dr. Pedder, and 12 months of then-current base salary in the case of Mr. Maness. |
| ● | Continuation of health insurance coverage for the Executive and their family for 18 months in the case of Dr. Roberts and Dr. Pedder, and 12 months in the case or Mr. Maness, or until the Executive becomes eligible for coverage under another employer’s plan. |
The Company’s obligation to provide such benefits is conditioned upon the Executive executing, and not revoking, a release of claims in a form acceptable to the Company.
Separation Benefits in Connection with a Change in Control. If the Company terminates the Executive’s employment without cause, or if the Executive resigns for good reason, in either case at the time of, or within six months following a change in control of the Company, then the Executive will be entitled to the following benefits:
| ● | 24 months of then-current base salary in the case of Dr. Roberts and Dr. Pedder, and 12 months of then- current base salary in the case of Mr. Maness. |
| ● | Accelerated vesting of all equity awards such that all awards are deemed to have been vested as of the date of termination. |
| ● | Continuation of health insurance coverage for the Executive and their family for 18 months in the case of Dr. Roberts and Dr. Pedder, and 12 months in the case or Mr. Maness, or until the Executive becomes eligible for coverage under another employer’s plan. |
The Company’s obligation to provide such benefits is conditioned upon the Executive executing, and not revoking, a release of claims in a form acceptable to the Company.
A “change in control” means the occurrence of any of the following events:
| ● | Any “person,” including a “group” (as such terms are used in Sections 13(d) and 14(d) of the Exchange Act) but excluding the Company, any entity controlling, controlled by or under common control with the Company, any trustee, fiduciary or other person or entity holding securities under any employee benefit plan or trust of the Company or any such entity, and, with respect to any particular qualified participant of any equity incentive plan of the Company (a “Participant”), the Participant and any “group” (as such term is used in Section 13(d)(3) of the Exchange Act) of which the Participant is a member), is or becomes the “beneficial owner” (as defined in Rule 13(d)(3) under the Exchange Act), directly or indirectly, of securities of the Company representing 50% or more of either (i) the combined voting power of the Company’s then-outstanding securities; or (ii) the Company’s then-outstanding equity securities (in either such case other than as a result of an acquisition of securities directly from the Company). |
| ● | Any consolidation or merger of the Company where the stockholders of the Company, immediately prior to the consolidation or merger, would not, immediately after the consolidation or merger, beneficially own, directly or indirectly, equity securities representing in the aggregate 50% or more of the combined voting power of the securities of the entity issuing cash or securities in the consolidation or merger (or of its ultimate parent entity, if any). |
| ● | Any sale, lease, exclusive license, exchange or other transfer (in one transaction or a series of transactions contemplated or arranged by any party as a single plan) of all or substantially all of the assets of the Company, other than a sale or disposition by the Company of all or substantially all of the Company’s assets to an entity, at least 50% of the combined voting power of the voting securities of which are owned by “persons” (as defined above) in substantially the same proportion as their ownership of the Company immediately prior to such sale. |
37
| ● | The members of the board at the beginning of any consecutive 24-calendar-month period (the “Incumbent Directors”) cease for any reason other than due to death to constitute at least a majority of the members of the board; provided that any director whose election, or nomination for election by the Company’s stockholders, was approved or ratified by a vote of at least a majority of the members of the board of directors then still in office who were members of the board of directors at the beginning of such 24-calendar-month period, will be deemed to be an Incumbent Director. |
Indemnification. The Company will indemnify each Executive and hold them harmless in connection with the defense of any lawsuit or other claim to which they are made a party by reason of being an officer, director, or employee of the Company, to the fullest extent permitted by Delaware law. This indemnification obligation does not extend to claims arising out of the Executives’ willful misconduct. In addition, the Company will use commercially reasonable efforts to maintain directors’ and officers’ liability insurance for each Executive for acts and omissions occurring during Executive’s employment with the Company.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The following table sets forth certain information with respect to the beneficial ownership of our Common Stock as of March 1, 2026 by:
| ● | Each of our named executive officers; | |
| ● | Each of our directors; |
| ● | All of our current directors and executive officers as a group; and |
| ● | Each person, or group of affiliated persons, who beneficially owned more than 5% of our Common Stock. |
We have determined beneficial ownership in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Except as indicated by the footnotes below, we believe, based on information furnished to us, that the persons and entities named in the table below have sole voting and sole investment power with respect to all shares of Common Stock that they beneficially owned, subject to applicable community property laws.
38
The percentage of shares beneficially owned is computed on the basis of 8,715,229 shares of Common Stock outstanding as of March 1, 2026. Shares of Common Stock that a person has the right to acquire within 60 days of March 1, 2026 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and executive officers as a group. Unless otherwise indicated, the address of each beneficial owner in the table below is 3540 Toringdon Way, Suite 200, #250 Charlotte, NC 28277.
| Name | Shares of Common Stock Beneficially Owned | Percentage of Common Stock Beneficially Owned | ||||||
| 5% Stockholders: | ||||||||
| Lucius Partners, LLC [1] | 3,626,026 | 39.88 | % | |||||
| Directors and Named Executive Officers: | ||||||||
| Michael J. Roberts, PhD[2] | 2,232,699 | 25.40 | % | |||||
| Simon C. Pedder, PhD[3] | 960,610 | 10.98 | % | |||||
| Timothy L. Maness, CPA[4] | 112,707 | 1.28 | ||||||
| L. Arthur Hewitt, PhD[5] | 18,308 | * | ||||||
| Patrick Gallagher[6] | 18,308 | * | ||||||
| Anthony Zook[7] | 18,308 | * | ||||||
| J. Nick Riehle[8] | 18,308 | * | ||||||
| All directors and officers as a group (7 persons) | 3,379,247 | 27.94 | % | |||||
| * | Less than 1%. |
| (1) | Includes 376,026 shares of our Common Stock issuable upon the exercise of immediately vested warrants the Placement Agent and/or its designees hold as of March 1, 2026. Matthew Eitner, the Chief Executive Officer of the Placement Agent, James Ahern, the Managing Partner of the Placement Agent, and Patrick Gallagher, a Managing Director of the Placement Agent, are managing members, members and/or officers of Lucius Partners. Mr. Eitner, but not Messrs. Ahern and Gallagher, has voting and investment control over securities held by the Placement Agent and Lucius Partners. The business address for Lucius Partners is 12 E. 49th Street, 11th Floor, New York, NY 10017. |
| (2) | Includes 73,231 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (3) | Includes 32,039 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (4) | Includes 112,707 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (5) | Includes 18,308 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (6) | Includes 18,308 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (7) | Includes 18,308 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
| (8) | Includes 18,308 common shares underlying outstanding options that are immediately exercisable or will be immediately exercisable within 60 days of March 1, 2026. |
39
Equity Compensation Plan Information
The following table provides information about the securities authorized for issuance under our equity compensation plan as of March 1, 2026.
| Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted- average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuances under equity compensation plans (excluding securities reflected in column (a)) | ||||||||||
| Equity compensation plans approved by securityholders: | ||||||||||||
| 2025 Equity Incentive Plan | 1,482,929 | (1) | $ | 4.40 | 455,539 | |||||||
| Equity compensation plans not approved by security holders: | ||||||||||||
| None | -- | -- | -- | |||||||||
| Total | 1,482,929 | $ | 4.40 | 455,539 | ||||||||
| (1) | The Company’s 2025 Equity Incentive Plan (the “2025 Plan”) was approved by Unite Acquisition stockholders prior to the Merger. For a period of ten years commencing on February 11, 2026, the share reserve for the 2025 Plan will be increased, subject to Board approval, by an amount equal to the lesser of (i) 4% of the number of shares outstanding as of December 31 of the immediately preceding calendar year or (ii) such lesser number of shares as determined by the Board. As of March 31, 2026, the Board had not yet approved any increase to the share reserve for 2026. |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Transactions with Lucius Partners and Related Persons
On February 11, 2025, we entered into an Advisory Services Agreement with Lucius Partners, and agreed to pay to Lucius Partners a cash fee of $180,000 for advisory services during the first year following the closing of our private placement offering on February 11, 2025, and agreed to pay Lucius Partners for advisory services, in advance for four consecutive three-month periods, commencing on the first day of the month that is the first full month 12 months or more after such closing, a cash fee of $45,000 for one year (such two-year period, the “Advisory Period”). The Advisory Period can be renewed for additional one-year periods upon written request by the Company within 60 days prior to the expiry of any Advisory Period.
Policies and Procedures for Related Person Transactions
Our Audit Committee has the primary responsibility for reviewing and approving or disapproving “related party transactions,” as defined in applicable SEC rules and regulations. As provided in the Audit Committee charter, in approving or rejecting any such transaction, our Audit Committee is to consider the relevant facts and circumstances available and deemed relevant to it, including, among other factors, whether the terms or other aspects of the transaction differ from those that would likely be negotiated with independent third parties.
All of the transactions described in this section were entered into prior to the adoption of the Audit Committee charter. Although we have not had a written policy for the review and approval of transactions with related persons, our board of directors has historically reviewed and approved any transaction where a director or officer had a financial interest, including the transactions described above. Prior to approving such a transaction, the material facts as to the relationship or interest of the relevant related person in the agreement or transaction were disclosed to our board of directors. Our board of directors took this information into account when evaluating the transaction and in determining whether such transaction was fair to us and in the best interest of all our stockholders.
40
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
The following table represents aggregate fees billed to the Company for the fiscal years ended December 31, 2025 and 2024, by WithumSmith+Brown PC (“Withum”), our principal accountant.
| Fiscal Year Ended | ||||||||
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Audit fees(1) | $ | 416,707 | $ | 119,600 | ||||
| Audit-related fees(2) | -- | -- | ||||||
| Tax fees(3) | -- | -- | ||||||
| All other fees(4) | -- | -- | ||||||
| Total | $ | 416,707 | $ | 119,600 | ||||
| (1) | Audit fees consist of the aggregate fees billed for professional services rendered for the audit of Adaptin Bio, Inc.’s annual financial statements and the reviews of the financial statements included in its Forms 10-Q and for any other services that were normally provided in connection with its statutory and regulatory filings or engagements. |
| (2) | Audit-related fees consist of fees for assurance and related services that are reasonably related to the performance of the Company’s audit or review of its financial statements. |
| (3) | Tax services principally include professional services rendered for tax compliance, tax advice and tax planning. |
| (4) | All other fees consist of fees for products and services provided, other than for the services reported under the headings “Audit Fees,” “Audit Related Fees” and “Tax Fees.” |
Audit Committee’s Pre-Approval Process
Our Audit Committee has adopted procedures requiring the pre-approval of all non-audit services performed by our independent registered public accounting firm in order to assure that these services do not impair the auditor’s independence. These procedures generally approve the performance of specific services subject to a cost limit for all such services. This general approval is reviewed, and if necessary modified, at least annually. Management must obtain the specific prior approval of the Audit Committee for each engagement of the independent registered public accounting firm to perform other audit-related or other non-audit services. The Audit Committee does not delegate its responsibility to approve services performed by the independent registered public accounting firm to any member of management. The standard applied by the Audit Committee in determining whether to grant approval of any type of non-audit service, or of any specific engagement to perform a non-audit service, is whether the services to be performed, the compensation to be paid therefore and other related factors are consistent with the independent registered public accounting firm’s independence under guidelines of the SEC and applicable professional standards. Relevant considerations include whether the work product is likely to be subject to, or implicated in, audit procedures during the audit of our financial statements, whether the independent registered public accounting firm would be functioning in the role of management or in an advocacy role, whether the independent registered public accounting firm’s performance of the service would enhance our ability to manage or control risk or improve audit quality, whether such performance would increase efficiency because of the independent registered public accounting firm’s familiarity with our business, personnel, culture, systems, risk profile and other factors, and whether the amount of fees involved, or the non-audit services portion of the total fees payable to the independent registered public accounting firm in the period would tend to reduce the independent registered public accounting firm’s ability to exercise independent judgment in performing the audit.
41
PART IV
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES.
(a)(1) Financial Statements
See Index to Consolidated Financial Statements on page F-1 of this Annual Report on Form 10-K, which is incorporated into this item by reference.
(a)(2) Financial Statement Schedules
All financial statement schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
(b) Exhibits
The following list of exhibits includes exhibits submitted with this Annual Report on Form 10-K as filed with the SEC and others incorporated by reference to other filings.
42
| + | Indicates a management contract or any compensatory plan, contract or arrangement. |
| * | Portions of this exhibit (indicated by asterisks) have been omitted in accordance with Item 601(b)(10) of Regulation S-K. The registrant hereby agrees to furnish supplementally copies of any of the omitted portions of this exhibit to the SEC upon its request. |
ITEM 16. FORM 10-K SUMMARY.
None.
43
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: April 1, 2026 | ADAPTIN BIO, INC. | |
| By: | /s/ Timothy L. Maness | |
| Name: | Timothy L. Maness | |
| Title: | Chief Financial Officer (On behalf of the Registrant and as Principal Financial and Accounting Officer) | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Position | Date | ||
| /s/ Michael J. Roberts | President, Chief Executive Officer and Director | April 1, 2026 | ||
| Michael J. Roberts | (Principal Executive Officer) | |||
| /s/ Simon C. Pedder | Executive Chairman and Director | April 1, 2026 | ||
| Simon C. Pedder | ||||
| /s/ Timothy L. Maness | Chief Financial Officer | April 1, 2026 | ||
| Timothy L. Maness | (Principal Financial and Accounting Officer) | |||
| /s/ Patrick Gallagher | Director | April 1, 2026 | ||
| Patrick Gallagher | ||||
| /s/ J. Nick Riehle | Director | April 1, 2026 | ||
| J. Nick Riehle | ||||
| /s/ Anthony Zook | Director | April 1, 2026 | ||
| Anthony Zook |
44